Čo je fenylketonúria
Tam fenylketonúria (P.K.U.) je to autozomálne recesívne dedičné metabolické ochorenie, ktoré postihuje 1 z 10 000 jedincov a zdá sa, že sa vyskytuje viac v homozygotnosti ako v heterozygotoch.
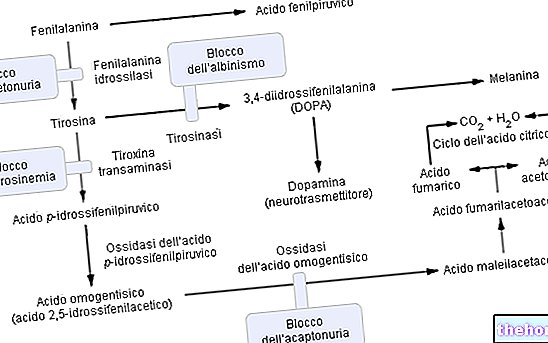
Fenylketonúria, ktorá patrí do skupiny hyperfenylalaninémie, významne ohrozuje metabolizmus fenylalanínu a najmä jeho konverzia na tyrozín; fenylketonúria je rozpoznaná zvýšenými hladinami fenylalanínu v moči a niektorých derivátov (fenylpyruvát, fenylacetát, fenylaktát a fenylacetylglutamín).
Najzávažnejšou komplikáciou fenylketonúrie je mentálne oneskorenie.
Fenylalanín, tyrozín a deriváty
Fenylalanín je esenciálna aminokyselina a tvorí väčšinu bielkovín v potrave; môže byť konvertovaný enzýmom fenylalanínhydroxyláza v tyrozíne (pridaním hydroxylovej skupiny -OH). Na druhej strane je tyrozín prekurzorovou aminokyselinou na syntézu:
- L-DOPA (medziprodukt syntézy dopamínu)
- Epinefrín
- Norepinefrín (všetky neurotransmitery).

Mechanizmus fenylketonúrie (P.K.U.)
Ako sa očakávalo, pri fenylketonúrii je v dôsledku jednej alebo viacerých (všetkých 6) chromozomálnych mutácií expresia (teda metabolická aktivita) fenylalanínhydroxylázy prakticky nulová. Tieto zmeny môžu byť rôzneho druhu (od zmien „missense“ po „zostrihové“ defekty alebo dokonca „čiastočné delécie“), ale dôležité je, že kvôli tejto enzymatickej neúčinnosti sú hladiny fenylalanínu v krvi (bežne 1 mg / 100 ml) v DOMINANTNEJ fenylketonúrii ľahko dosiahnu množstvo dokonca 50 -krát vyššie.
Fungovanie enzýmu fenylalanínhydroxylázy: Na výrobu tyrozínu (+ dihydrobiopterínu) vyžaduje fenylalanínhydroxyláza: fenylalanín, kyslík a tetrahydrobiopterín (redukovaný pteridín, ktorý funguje ako koofaktor); reakcia je tiež reverzibilná a dihydrobiopterín je možné znova premeniť (vďaka enzýmu dihydropterín reduktáza) v tetrahydrobiopteríne.
Komplikácie
Fenylketonúria môže spôsobiť viac alebo menej závažné komplikácie na základe závažnosti patologického prejavu a včasnosti diagnostiky; Ako dedičnú patológiu sa fenylketonúria rozlišuje v:
- Dominantný, preto charakterizovaný KOMPLETNOU neaktivitou enzýmu fenylalanínhydroxylázy
- Recesívne, v ktorom je aktívnych iba 30% z celkového enzymatického dedičstva.
Komplikácie fenylketonúrie možno pripísať, a priamo úmerné, metabolickej akumulácii fenylalanínu, jeho derivátov a zníženej syntéze tyrozínu. V patológii je prebytočný fenylalanín relatívne efektívne filtrovaný obličkami, ktoré ho len čiastočne reabsorbujú a vylučujú močom Perzistencia hladín hyperfenylalaninémie však určuje metabolickú reakciu molekulárneho KONVERZIE v kyselina fenylpyrohroznová a / alebo iné deriváty, ktoré sa ľahšie odvádzajú (fenylpyruvát, fenylacetát, fenylaktát).
Fenylketonúria komplikuje toxicita fenylalanínu, kyseliny fenylpyruvovej a jej derivátov voči centrálnemu nervovému systému (CNS). Ich nadmerná prítomnosť vo vývoji mozgu neúprosne určuje formu mentálnej retardácie.
Pozn. Plazmatické koncentrácie ostatných aminokyselín sú mierne znížené, pravdepodobne v dôsledku spätnej väzby o intestinálnej absorpcii alebo renálnej tubulárnej reabsorpcii.
Poškodenie mozgu, ako závažná komplikácia fenylketonúrie, je spôsobené odčítaním iných esenciálnych aminokyselín v proteosyntéze, najmä pri tvorbe polyribozómov, myelínu, noradrenalínu a serotonínu. Fenylketonúria - nie je viditeľná bezprostredne po narodení, ale po niekoľkých rokoch - ak nie je liečená, vyžaduje hospitalizáciu dieťaťa a je úplne nevratná.
Pokročilá fenylketonúria môže byť tiež dobre viditeľná voľným okom; vysoké koncentrácie fenylalanínu, ktoré inhibujú enzým tyrozináza, významne zhoršujú syntézu melanínu znížením pigmentácie pokožky a vlasov; okrem toho akumulácia fenylacetátu vo vlasoch a koži dodáva fenylketonurikom silný a nepríjemný „myšací zápach“.












-cos-cause-e-disturbi-associati.jpg)