Všeobecnosť
Mitochondriálna DNA alebo mtDNA je deoxyribonukleová kyselina, ktorá sídli vo vnútri mitochondrií, tj. Organel eukaryotických buniek zodpovedných za veľmi dôležitý bunkový proces oxidačnej fosforylácie.

Má však aj niektoré zvláštnosti, štrukturálne i funkčné, vďaka ktorým je jedinečný svojho druhu. Medzi tieto zvláštnosti patrí: kruhovitosť dvojvlákna nukleotidov, obsah génov (čo je iba 37 prvkov) a takmer úplná absencia nekódujúcich nukleotidových sekvencií.
Mitochondriálna DNA plní základnú funkciu pre prežitie buniek: produkuje enzýmy potrebné na realizáciu oxidačnej fosforylácie.
Čo je mitochondriálna DNA?
Mitochondriálna DNA alebo mtDNA je DNA umiestnená v mitochondriách.
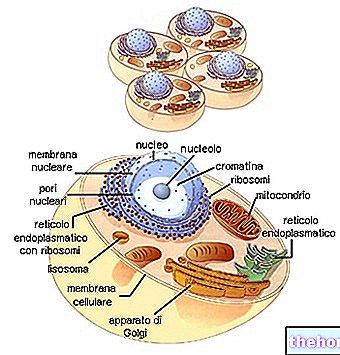
Mitochondrie sú veľké bunkové organely, typické pre eukaryotické organizmy, ktoré premieňajú chemickú energiu obsiahnutú v potravinách na ATP, čo je forma energie, ktorú môžu bunky využívať.
POZADIE ŠTRUKTÚRY A FUNKCIE MITOCHONDRÓNOV
Mitochondrie sú tubulárne, vláknité alebo zrnitého tvaru a nachádzajú sa v cytoplazme, ktorá zaberá takmer 25% jej objemu.
Majú dve fosfolipidové dvojvrstvové membrány, jednu vonkajšiu a jednu vnútornú.
Vonkajšia membrána, známa ako vonkajšia mitochondriálna membrána, predstavuje obvod každého mitochondrií a má transportné proteíny (poriny a ďalšie), vďaka ktorým je priepustná pre molekuly s veľkosťou rovnou alebo menšou ako 5 000 daltonov.
Vnútorná membrána, známa ako vnútorná mitochondriálna membrána, obsahuje všetky enzymatické (alebo enzymatické) a koenzýmové zložky potrebné na syntézu ATP a definuje centrálny priestor nazývaný matrica.
Na rozdiel od vonkajšej membrány má vnútorná mitochondriálna membrána početné invaginácie - takzvané hrebene - ktoré zvyšujú jej celkovú plochu.
Medzi dvoma mitochondriálnymi membránami je priestor takmer 60-80 angstrômov (A). Tento priestor sa nazýva intermembránový priestor. Intermembránový priestor má zloženie veľmi podobné zloženiu cytoplazmy.
Syntéza ATP, ktorú prevádzkujú mitochondrie, je veľmi zložitý proces, ktorý biológovia stotožňujú s pojmom oxidačná fosforylácia.
PRESNÉ UMIESTNENIE MITOCHONDRÁLNEJ DNA A MNOŽSTVA

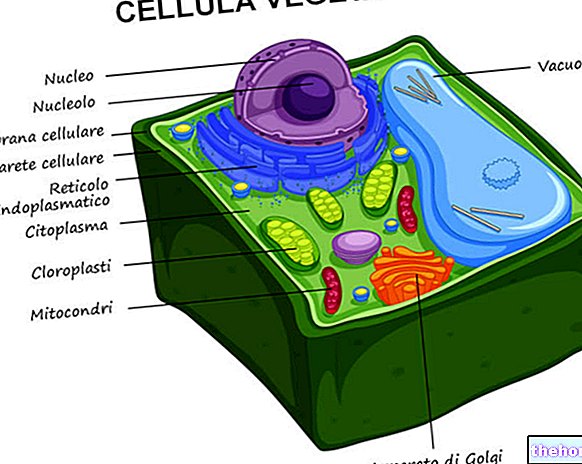
Obrázok: ľudská mitochondria.
Mitochondriálna DNA sídli v mitochondriálnej matrici, t.j. v priestore vymedzenom vnútornou mitochondriálnou membránou.
Podľa spoľahlivých vedeckých štúdií môže každý mitochondrión obsahovať od 2 do 12 kópií mitochondriálnej DNA.
Vzhľadom na skutočnosť, že v ľudskom tele môžu niektoré bunky obsahovať niekoľko tisíc mitochondrií, môže celkový počet kópií mitochondriálnej DNA v jednej ľudskej bunke dosiahnuť až 20 000 jednotiek.
Vezmite prosím na vedomie: počet mitochondrií v ľudských bunkách sa líši v závislosti od typu bunky. Napríklad hepatocyty (t.j. pečeňové bunky) môžu obsahovať 1 000 až 2 000 mitochondrií, zatiaľ čo erytrocyty (t.j. červené krvinky) ich úplne neobsahujú.
Štruktúra
Celková štruktúra mitochondriálnej molekuly DNA sa podobá všeobecnej štruktúre jadrovej DNA, to znamená genetickému dedičstvu prítomnému v jadre eukaryotických buniek.
Naozaj, analogicky k jadrovej DNA:
- Mitochondriálna DNA je biopolymér pozostávajúci z dvoch dlhých reťazcov nukleotidov. Nukleotidy sú organické molekuly, ktoré sú výsledkom spojenia troch prvkov: cukru s 5 atómami uhlíka (v prípade DNA deoxyribózy), dusíkatej bázy a fosfátovej skupiny.
- Každý nukleotid mitochondriálnej DNA sa viaže na nasledujúci nukleotid toho istého vlákna pomocou fosfodiesterovej väzby medzi uhlíkom 3 jeho deoxyribózy a fosfátovou skupinou bezprostredne nasledujúceho nukleotidu.
- Dva vlákna mitochondriálnej DNA majú opačnú orientáciu, pričom koniec jedného interaguje s koncom druhého a naopak.Toto konkrétne usporiadanie je známe ako antiparalelné usporiadanie (alebo antiparalelná orientácia).
- Tieto dva vlákna mitochondriálnej DNA na seba vzájomne pôsobia prostredníctvom dusíkatých báz.
Konkrétne každá dusíkatá báza každého vlákna vytvára vodíkové väzby s jednou a iba jednou dusíkatou zásadou prítomnou na druhom vlákne.
Tento typ interakcie sa nazýva „párovanie dusíkatých báz“ alebo „pár dusíkatých báz“. - Dusíkaté bázy mitochondriálnej DNA sú adenín, tymín, cytozín a guanín.
Párovanie, z ktorého vznikajú tieto dusíkaté bázy, nie je náhodné, ale je veľmi špecifické: adenín interaguje iba s tymínom, zatiaľ čo cytozín interaguje iba s guanínom. - Mitochondriálna DNA je domovom génov (alebo génových sekvencií). Gény sú sekvencie viac alebo menej dlhých nukleotidov s dobre definovaným biologickým významom. Vo väčšine prípadov vedú k vzniku bielkovín.

ŠTRUKTURÁLNE ÚDAJE MITOCHONDRÁLNEJ DNA
Okrem vyššie uvedených analógií má ľudská mitochondriálna DNA niektoré štrukturálne zvláštnosti, ktoré ju výrazne odlišujú od ľudskej jadrovej DNA.
Po prvé, je to kruhová molekula, zatiaľ čo jadrová DNA je lineárna molekula.
Má teda 16 569 párov dusíkatých báz, zatiaľ čo jadrová DNA má obrovských 3,3 miliardy.
Obsahuje 37 génov, zatiaľ čo jadrová DNA obsahuje asi 20 000 až 25 000.
Nie je organizovaná v chromozómoch, zatiaľ čo jadrová DNA je rozdelená na 23 chromozómov a tvorí s niektorými špecifickými proteínmi látku nazývanú chromatín.
Nakoniec obsahuje sériu nukleotidov, ktoré sa zúčastňujú dvoch génov súčasne, zatiaľ čo jadrová DNA má gény, ktorých nukleotidové sekvencie sú dobre definované a navzájom odlišné.
Pôvod
Mitochondriálna DNA má s najväčšou pravdepodobnosťou „bakteriálny“ pôvod.
Na základe mnohých nezávislých štúdií sa molekulárni biológovia domnievajú, že bunková prítomnosť mitochondriálnej DNA je výsledkom začlenenia nezávislých bakteriálnych organizmov, predných eukaryotických buniek, predkovými eukaryotickými bunkami, veľmi podobných mitochondriám.
Tento kuriózny objav len čiastočne ohromil vedeckú komunitu, pretože DNA prítomná v baktériách je spravidla kruhový nukleotidový reťazec, podobne ako mitochondriálna DNA.
Teória, podľa ktorej majú mitochondrie a mitochondriálna DNA „bakteriálny pôvod“, dostala názov „endosymbiotická teória“ od slova „endosymbióza“. Stručne povedané, v biológii termín „endosymbióza“ označuje spoluprácu medzi dvoma organizmami, ktorá zahŕňa „zapracovanie jedného do druhého, s cieľom získať určitú výhodu.
Zvedavosť
Podľa spoľahlivých vedeckých štúdií by v priebehu evolúcie mnohé bakteriálne gény prítomné v budúcej mitochondriálnej DNA zmenili umiestnenie a presťahovali by sa do jadrovej DNA.
Inými slovami, na začiatku endosymbiózy niektoré gény teraz prítomné v jadrovej DNA sídlili v DNA týchto bakteriálnych organizmov, z ktorých sa neskôr stali mitochondrie.
Na podporu teórie týkajúcej sa posunu génov medzi mitochondriálnou DNA a jadrovou DNA je pozorovanie, že niektoré gény pochádzajú z mitochondriálnej DNA v niektorých druhoch a z jadrovej DNA v iných.
Funkcia
Mitochondriálna DNA produkuje enzýmy (tj. Proteíny), nevyhnutné pre správnu implementáciu delikátneho procesu oxidatívnej fosforylácie.
Pokyny na syntézu týchto enzýmov sa nachádzajú v 37 génoch, ktoré tvoria genóm mitochondriálnej DNA.
ČO KÓDY MITOCHONDRÁLNYCH DNA GÉNOV: PODROBNOSTI
37 génov mitochondriálnej DNA kóduje: proteíny, tRNA a rRNA.
Najmä:
- 13 kóduje 13 proteínov zodpovedných za oxidačnú fosforyláciu
- 22 kód pre 22 molekúl tRNA
- 2 kódujú 2 molekuly rRNA
Molekuly tRNA a rRNA sú zásadné pre syntézu vyššie uvedených 13 proteínov, pretože tvoria mechanizmus, ktorý reguluje ich produkciu.
Inými slovami, mitochondriálna DNA obsahuje informácie na výrobu určitého súboru bielkovín a nástrojov potrebných na ich syntézu.
Čo sú RNA, tRNA a rRNA?
RNA alebo ribonukleová kyselina je nukleová kyselina, ktorá hrá zásadnú úlohu pri vytváraní bielkovín, počínajúc DNA.
Spravidla je ANN jednovláknový, môže existovať v rôznych formách (alebo typoch) v závislosti od konkrétnej funkcie, na ktorú je delegovaný.
TRNA a rRNA sú dve z týchto možných foriem.
TRNA sa používa na pridanie aminokyselín počas procesu výroby bielkovín. Aminokyseliny sú molekulárne jednotky, ktoré tvoria proteíny.
RRNA tvorí ribozómy, to sú bunkové štruktúry, v ktorých prebieha syntéza bielkovín.
Ak chcete podrobne poznať ANN a jeho funkcie, čitatelia môžu kliknúť sem.
FUNKČNÉ PODROBNOSTI O MITOCHONDRÁLNEJ DNA
Z funkčného hľadiska má mitochondriálna DNA niektoré zvláštne vlastnosti, ktoré ju jasne odlišujú od jadrovej DNA.
Z čoho pozostávajú tieto zvláštne vlastnosti:
- Mitochondriálna DNA je polonezávislá v tom zmysle, že potrebuje zásah niektorých proteínov syntetizovaných z jadrovej DNA.
Na druhej strane je jadrová DNA úplne autonómna a sama produkuje všetko, čo potrebuje na správne plnenie svojich úloh. - Mitochondriálna DNA má mierne odlišný genetický kód ako jadrová DNA. To vedie k množstvu rozdielov pri tvorbe bielkovín: ak určitá sekvencia nukleotidov v jadrovej DNA vedie k vytvoreniu určitého proteínu, rovnaká sekvencia v mitochondriálnej DNA vedie k vzniku mierne odlišného proteínu.
- Mitochondriálna DNA má veľmi málo nekódujúcich nukleotidových sekvencií, to znamená, že neprodukujú žiadne proteíny, tRNA ani rRNA. V percentuálnom vyjadrení sú nekódujúce iba 3% mitochondriálnej DNA.
Na druhej strane jadrová DNA kóduje iba 7%, takže obsahuje veľa nekódujúcich nukleotidových sekvencií (až 93%).
Tabuľka: súhrn rozdielov medzi ľudskou mitochondriálnou DNA a ľudskou jadrovou DNA.
Mitochondriálna DNA
Jadrová DNA
- Je kruhový
- Je lineárny
- Má celkom 16 569 párov dusíkatých báz
- Má celkom 3,3 miliardy párov dusíkatých báz
- Obsahuje celkom 37 génov
- Obsahuje 20 000 až 25 000 génov
- Na správnu funkciu potrebuje podporu niektorých génových produktov pochádzajúcich z jadrovej DNA
- Je autonómny a sám produkuje všetko, čo potrebuje na správne plnenie svojich funkcií
- Môže byť prítomný v niekoľkých kópiách v rámci každej jednotlivej mitochondrie
- Je jedinečný, to znamená, že je iba v jednej kópii a nachádza sa v jadre
- Kóduje 97% nukleotidovej sekvencie, ktorá ju tvorí
- Kóduje iba 7% nukleotidovej sekvencie, ktorá ju tvorí
- Nie je organizovaný do chromozómov
- Je rozdelená na 23 chromozómov
- Používa genetický kód mierne odlišný od takpovediac „tradičného“ kódu
- Použite „tradičný“ genetický kód
- Jeho dedičstvo je materské
- Jeho dedičstvo je napoly materské a napoly otcovské
- Niektoré z jeho nukleotidov sa zúčastňujú dvoch génov súčasne
- Sekvencie nukleotidov, ktoré tvoria gény, sú navzájom dobre odlíšené
Dedičnosť
Dedičnosť mitochondriálnej DNA je výlučne materská.
To znamená, že v páre rodičov je to žena, ktorá prenáša mitochondriálnu DNA na potomstvo (tj. Na deti).
Úplne opačným spôsobom ako vyššie uvedené je dedičnosť jadrovej DNA z polovice materská a z polovice otcovská. Inými slovami, obaja rodičia prispievajú rovnako k prenosu jadrovej DNA u potomstva.
Vezmite prosím na vedomie: materská dedičnosť mitochondriálnej DNA zahŕňa aj mitochondriálnu štruktúru. Mitochondrie prítomné u jednotlivca sú teda materské.
Súvisiace patológie
Predpoklad: Genetická mutácia je trvalá zmena v sekvencii nukleotidov, ktoré tvoria jadrový alebo mitochondriálny gén DNA.
Prítomnosť genetickej mutácie má spravidla za následok „zmenu alebo stratu normálnej funkcie zapojeného génu.
Prítomnosť mutácií v mitochondriálnych génoch DNA môže viesť k širokému spektru chorôb, vrátane:
- Leberova dedičná optická neuropatia
- Kearns-Sayreov syndróm
- Leighov syndróm
- Nedostatok oxidázy cytochrómu C.
- Progresívna vonkajšia oftalmoplegia
- Pearsonov syndróm
- Mitochondriálna encefalomyopatia s laktátovou acidózou a epizódami podobnými mŕtvici (syndróm MELAS)
- Cukrovka s materskou hluchotou
- Myoklonická epilepsia s nepravidelnými červenými vláknami
Pokiaľ ide o patologické stavy súvisiace s jednou alebo viacerými mitochondriálnymi mutáciami DNA, je potrebné objasniť dva aspekty.
Po prvé, závažnosť ochorenia závisí od kvantitatívneho vzťahu medzi mutovanými mitochondriálnymi DNA a zdravými, normálnymi mitochondriálnymi DNA. Ak je počet mutovaných mitochondriálnych DNA výrazne vyšší ako počet zdravých DNA, bude výsledný stav vážnejší.
Za druhé, mutácie v mitochondriálnej DNA postihujú iba niektoré tkanivá organizmu, najmä tie, ktoré vyžadujú veľké množstvo ATP vyplývajúce z procesu oxidačnej fosforylácie. To je celkom pochopiteľné: bunky, ktoré najviac potrebujú, trpieť viac ako jednou poruchou mitochondriálnej DNA funkciu, ktorú mitochondriálna DNA normálne plní.
LEBEROVA HEREDITÁRNA OPTICKÁ NEUROPATIA
Leberova dedičná optická neuropatia vzniká v dôsledku mutácie až štyroch mitochondriálnych génov DNA. Tieto gény obsahujú informácie, ktoré vedú k syntéze takzvaného komplexu I (alebo NADH oxid-reduktázy), jedného z rôznych enzýmov zapojených do procesu oxidačnej fosforylácie.
Prejavy patológie spočívajú v postupnej degenerácii zrakového nervu a postupnej strate zraku.
KEARNS-SAYRE SYNDROM
Kearns-Sayreov syndróm sa objavuje v dôsledku nedostatku primeraného množstva mitochondriálnej DNA (Poznámka: nedostatok určitej nukleotidovej sekvencie sa nazýva delécia).
U ľudí s Kearns-Sayreovým syndrómom sa vyvíja oftalmoplegia (celková alebo čiastočná paralýza okulomotorických svalov), forma retinopatie a abnormalít srdcového rytmu (atrioventrikulárna blokáda).
LEIGHOV SYNDROM
Leighov syndróm vzniká ako dôsledok mitochondriálnych mutácií DNA, ktoré môžu ovplyvniť proteín ATP-syntázu (tiež nazývaný V-komplex) a / alebo niektoré tRNA.
Leighov syndróm je progresívne neurologické ochorenie, ktoré sa objavuje v detstve alebo v detstve a je zodpovedné za: oneskorenie vývoja, svalovú slabosť, periférnu neuropatiu, motorické poruchy, dýchacie ťažkosti a oftalmoplegiu.
ZÁVADA CYTOCHROMOVEJ OXIDÁZY
Nedostatok oxidázy cytochrómu C nastáva v dôsledku mutácie najmenej 3 mitochondriálnych génov DNA. Tieto gény sú nevyhnutné pre správnu syntézu enzýmu oxidázy cytochrómu C (alebo komplexu IV), zapojeného do procesu oxidačnej fosforylácie.
Typické prejavy nedostatku oxidázy cytochrómu C pozostávajú z: dysfunkcie kostrového svalstva, srdcovej dysfunkcie, renálnej dysfunkcie a hepatálnej dysfunkcie.
PROGRESÍVNA VONKAJŠIA OFTALMOPLEGIA
Progresívna vonkajšia oftalmoplegia vzniká z nedostatku podstatného počtu mitochondriálnych DNA nukleotidov (delécia)
S progresívnym charakterom (ako sa dá odhadnúť z názvu) spôsobuje táto patológia paralýzu okulomotorických svalov s následnou ptózou a značnými problémami so zrakom.
PEARSONOV SYNDROM
Pearsonov syndróm sa objavuje po nápadnom odstránení mitochondriálnej DNA podobným spôsobom ako progresívna vonkajšia oftalmoplegia a Kearns-Sayreov syndróm.
Typickými prejavmi Pearsonovho syndrómu sú: sideroblastická anémia, pankreatická dysfunkcia (napr. Diabetes závislý od inzulínu), neurologické deficity a svalové poruchy.
Pearsonov syndróm spravidla spôsobuje, že postihnutý v mladom veku zomrie. V skutočnosti ľudia postihnutí touto patológiou len zriedka dosiahnu dospelosť.
MELASOV SYNDROM
Syndróm MELAS, tiež známy ako mitochondriálna encefalomyopatia s laktátovou acidózou a epizódami podobnými mŕtvici, vzniká mutáciou najmenej 5 mitochondriálnych génov DNA.
Tieto gény prispievajú k syntéze NADH oxid reduktázy alebo komplexu I a niektorých tRNA.
Syndróm MELAS zahŕňa prítomnosť neurologických porúch, svalových porúch, neobvyklého hromadenia kyseliny mliečnej v tkanivách (so všetkými sprievodnými príznakmi), problémy s dýchaním, stratu kontroly funkcie čriev, opakujúcu sa únavu, problémy s obličkami, srdcové problémy, cukrovku, epilepsiu a nedostatok koordinácie.
INÉ PATOLÓGIE
Podľa rôznych vedeckých štúdií by choroby, ako je syndróm cyklického vracania, retinitis pigmentosa, ataxia, Parkinsonova choroba a Alzheimerova choroba, tiež videli zapojenie mitochondriálnej DNA a niektorých jej mutácií.



-cos-e-malattia-nelluomo.jpg)