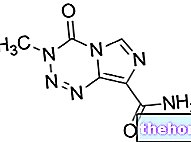
Účinné látky: Abiraterón (Abiraterón acetát)
ZYTIGA 250 mg tablety
Prečo sa používa Zytiga? Načo to je?
ZYTIGA obsahuje liek nazývaný abiraterón acetát. Používa sa na liečbu rakoviny prostaty u dospelých mužov, ktorá sa rozšírila do iných častí tela. ZYTIGA zastavuje v tele tvorbu testosterónu, ktorý môže spomaliť rast rakoviny prostaty.
Keď užívate tento liek, lekár vám predpíše aj iný liek nazývaný prednizón alebo prednizolón. Tento liek sa používa na zníženie možnosti vysokého krvného tlaku, príliš veľkého množstva vody v tele (zadržiavanie tekutín) alebo nízkych hladín chemikálie známej ako draslík v krvi.
Kontraindikácie Kedy by sa nemal používať Zytiga
Neužívajte ZYTIGU
- ak ste alergický na abiraterón acetát alebo na ktorúkoľvek z ďalších zložiek tohto lieku (uvedených v časti 6).
- ak ste žena, najmä ak ste tehotná. Použitie ZYTIGY je určené len pre mužov.
- ak máte závažné poškodenie pečene.
Neužívajte tento liek, ak sa vás niečo z uvedeného týka. Ak si nie ste niečím istý, opýtajte sa svojho lekára alebo lekárnika predtým, ako začnete užívať tento liek.
Opatrenia pri používaní Čo potrebujete vedieť predtým, ako užijete Zytigu
Predtým, ako začnete užívať tento liek, obráťte sa na svojho lekára alebo lekárnika:
- ak máte problémy s pečeňou
- ak vám bolo povedané, že máte vysoký krvný tlak alebo srdcové zlyhanie alebo nízku hladinu draslíka v krvi (nízka hladina draslíka môže zvýšiť riziko problémov so srdcovým rytmom)
- ak ste niekedy mali iné problémy so srdcom alebo cievami
- ak máte nepravidelný alebo rýchly srdcový tep
- ak dýchate
- ak ste rýchlo pribrali
- ak máte opuch nôh, členkov alebo nôh
- ak ste v minulosti užívali liek nazývaný ketokonazol na liečbu rakoviny prostaty
- o potrebe užívať tento liek s prednizónom alebo prednizolónom
- o možných účinkoch na kosti
- ak máte vysokú hladinu cukru v krvi.
Povedzte svojmu lekárovi, ak vám povedali, že máte akékoľvek problémy so srdcom alebo cievami, vrátane problémov so srdcovým rytmom (arytmia) alebo ak sa liečite liekmi na tieto stavy.
Ak máte žltú pokožku alebo oči, tmavý moč alebo silnú nevoľnosť alebo vracanie, povedzte to svojmu lekárovi, pretože to môžu byť prejavy alebo príznaky problémov s pečeňou. Zriedkavo sa môže vyskytnúť problém s funkciou pečene (nazývaný akútne zlyhanie pečene), ktorý môže viesť k smrti.
Môže sa vyskytnúť znížený počet červených krviniek, znížená sexuálna túžba (libido), svalová slabosť a / alebo bolesť svalov.
Ak si nie ste istý, či sa vás týka niektorý z vyššie uvedených bodov, porozprávajte sa so svojím lekárom alebo lekárnikom predtým, ako začnete užívať tento liek.
Monitorovanie krvi
ZYTIGA môže postihnúť pečeň a nemusí mať žiadne príznaky. Keď užívate tento liek, váš lekár vám bude pravidelne kontrolovať krv, aby zistil, či nemá liek ZYTIGA vplyv na pečeň.
Deti a dospievajúci
Liek nie je indikovaný u detí a dospievajúcich. Ak dieťa ZYTIGA náhodne prehltne dieťa alebo mladistvý, okamžite choďte do nemocnice a vezmite si so sebou písomnú informáciu pre používateľov, aby ste to ukázali lekárovi na pohotovosti.
Interakcie Ktoré lieky alebo potraviny môžu zmeniť účinok Zytigy
Predtým, ako začnete užívať akýkoľvek liek, opýtajte sa svojho lekára alebo lekárnika.
Ak teraz užívate alebo ste v poslednom čase užívali, či práve budete užívať ďalšie lieky, povedzte to svojmu lekárovi alebo lekárnikovi. Je to dôležité, pretože ZYTIGA môže zvýšiť účinok niektorých liekov vrátane liekov na srdce, trankvilizérov, liekov z liečivých bylín (napr. Ľubovník bodkovaný) a iných. Váš lekár sa môže rozhodnúť zmeniť dávku týchto liekov. Niektoré lieky môžu tiež zvýšiť alebo znížiť účinky ZYTIGY, ktoré môžu viesť k vedľajším účinkom alebo ZYTIGA nemusia fungovať tak, ako by mali.
Iné lieky užívané spolu so ZYTIGOU
Liečba depriváciou androgénu môže zvýšiť riziko problémov so srdcovým rytmom.
Ak užívate nejaké lieky, povedzte to svojmu lekárovi:
- používané na liečbu problémov so srdcovým rytmom (napr. chinidín, prokainamid, amiodarón a sotalol);
- je známe, že zvyšuje riziko problémov so srdcovým rytmom [napr. metadón (používa sa na úľavu od bolesti a na liečbu drogovej závislosti), moxifloxacín (antibiotikum), antipsychotiká (používa sa pri ťažkých duševných chorobách)].
ZYTIGA s jedlom
- Tento liek sa nesmie užívať s jedlom (pozri časť „Užívanie tohto lieku“).
- Užívanie ZYTIGY s jedlom môže spôsobiť vedľajšie účinky.
Upozornenia Je dôležité vedieť, že:
Tehotenstvo a dojčenie
Použitie ZYTIGY nie je indikované u žien.
- Tento liek môže spôsobiť poškodenie plodu, ak ho užívajú tehotné ženy.
- Tehotné ženy alebo ženy, ktoré môžu byť tehotné, by mali používať rukavice, ak sa potrebujú dotknúť lieku ZYTIGA alebo s ním zaobchádzať.
- Ak máte sex so ženou v plodnom veku, mali by ste použiť kondóm a iné „účinné antikoncepčné opatrenie. Ak máte sex s tehotnou ženou, použite kondóm na ochranu plodu“.
Vedenie vozidla a obsluha strojov
Tento liek pravdepodobne neovplyvní vašu schopnosť viesť vozidlo a obsluhovať akékoľvek nástroje alebo stroje.
ZYTIGA obsahuje laktózu a sodík
- ZYTIGA obsahuje laktózu (druh cukru). Ak vám váš lekár povedal, že neznášate niektoré cukry, kontaktujte svojho lekára pred užitím tohto lieku.
- Tento liek obsahuje približne 27 mg sodíka v dennej dávke štyroch tabliet. Toto je potrebné vziať do úvahy u pacientov na diéte so zníženým obsahom sodíka.
Dávka, spôsob a čas podávania Ako používať Zytigu: Dávkovanie
Vždy užívajte tento liek presne tak, ako vám povedal váš lekár. Ak si nie ste niečím istý, obráťte sa na svojho lekára alebo lekárnika.
Koľko vziať
Odporúčaná dávka je 1 000 mg (štyri tablety) jedenkrát denne.
Užívanie tohto lieku
- Užívajte tento liek ústami.
- Neužívajte ZYTIGU s jedlom.
- Užívajte ZYTIGU najmenej dve hodiny po jedle a najmenej jednu hodinu po užití ZYTIGY nič nejedzte (pozri časť 2 „ZYTIGA s jedlom“).
- Tablety prehltnite celé a zapite trochou vody.
- Tablety nelámte.
- ZYTIGA sa užíva s liekom nazývaným prednizón alebo prednizolón. Užívajte prednizón alebo prednizolón presne podľa pokynov lekára.
- Počas užívania ZYTIGY musíte užívať prednizón alebo prednizolón každý deň.
- V prípade núdze môže byť potrebné zmeniť množstvo prednizónu alebo prednizolónu. Váš lekár vám poradí, ak potrebujete zmeniť množstvo prednizónu alebo prednizolónu, ktoré užívate. Neprestaňte užívať prednizón alebo prednizolón, pokiaľ vám to nepovie váš lekár.
Váš lekár vám môže predpísať aj iné lieky počas užívania ZYTIGY a prednizónu alebo prednizolónu.
Ak zabudnete užiť ZYTIGU
- Ak zabudnete užiť ZYTIGU alebo prednizón alebo prednizolón, užite zvyčajnú dávku nasledujúci deň.
- Ak zabudnete užiť ZYTIGU alebo prednizón alebo prednizolón na viac ako jeden deň, porozprávajte sa so svojím lekárom bez toho, aby ste museli dlho čakať.
Ak prestanete užívať ZYTIGU
Neprestaňte užívať ZYTIGU alebo prednizón alebo prednizolón, pokiaľ vám to nepovie váš lekár.
Ak máte ďalšie otázky týkajúce sa použitia tohto lieku, opýtajte sa svojho lekára alebo lekárnika.
Predávkovanie Čo robiť, ak ste užili príliš veľa Zytigy
Ak užijete viac ZYTIGY, ako máte, ihneď sa poraďte so svojím lekárom alebo choďte do nemocnice.
Vedľajšie účinky Aké sú vedľajšie účinky lieku Zytiga
Tak ako všetky lieky, aj tento liek môže spôsobovať vedľajšie účinky, hoci sa neprejavia u každého.
Ak spozorujete niektorý z vedľajších účinkov uvedených v tejto písomnej informácii pre používateľov, prestaňte užívať ZYTIGU a ihneď kontaktujte lekára:
- svalová slabosť, svalové kŕče alebo pocit búšenia srdca (palpitácie). Môžu to byť príznaky nízkej hladiny draslíka v krvi.
Ďalšie vedľajšie účinky zahŕňajú:
Veľmi časté (môžu postihnúť viac ako 1 z 10 pacientov)
Tekutina v nohách alebo chodidlách, nízka hladina draslíka v krvi, vysoký krvný tlak, infekcia močových ciest, hnačka.
Časté (môžu postihnúť až 1 z 10 pacientov)
Vysoká hladina tuku v krvi, zvýšené pečeňové testy, bolesť na hrudníku, poruchy srdcového rytmu, srdcové zlyhanie, rýchly srdcový rytmus, závažná infekcia nazývaná sepsa, zlomeniny kostí, poruchy trávenia, krv v moči, vyrážka.
Menej časté (môžu postihnúť až 1 zo 100 pacientov)
Problémy s nadobličkami (súvisiace s problémami so soľou a vodou), svalová slabosť a / alebo bolesť svalov.
Zriedkavé (môžu postihnúť až 1 z 1 000 pacientov)
Podráždenie pľúc (tiež nazývaná alergická alveolitída). Problémy s funkciou pečene (nazývané tiež akútne zlyhanie pečene).
Neznáme (frekvenciu nemožno odhadnúť z dostupných údajov)
Infarkt, zmeny na EKG - elektrokardiogram (predĺženie QT).
K úbytku kostnej hmoty môže dôjsť u mužov liečených na rakovinu prostaty. ZYTIGA v kombinácii s prednizónom alebo prednizolónom môže zvýšiť stratu kostnej hmoty.
Hlásenie vedľajších účinkov
Ak sa u vás vyskytne akýkoľvek vedľajší účinok, obráťte sa na svojho lekára alebo lekárnika. To sa týka aj akýchkoľvek vedľajších účinkov, ktoré nie sú uvedené v tejto písomnej informácii. Vedľajšie účinky môžete hlásiť aj priamo prostredníctvom národného systému hlásenia uvedeného v Prílohe V. Hlásením vedľajších účinkov môžete prispieť k získaniu ďalších informácií o bezpečnosti tohto lieku.
Expirácia a retencia
- Tento liek uchovávajte mimo dohľadu a dosahu detí.
- Nepoužívajte tento liek po dátume exspirácie, ktorý je uvedený na škatuli a fľaši. Dátum exspirácie sa vzťahuje na posledný deň v tomto mesiaci.
- Uchovávajte pri teplote do 30 ° C.
- Nevyhadzujte žiadne lieky do vody alebo domového odpadu. Opýtajte sa svojho lekárnika, ako zlikvidovať lieky, ktoré už nepoužívate. Pomôže to chrániť životné prostredie.
Termín "> Ďalšie informácie
Čo ZYTIGA obsahuje
- Účinnou zložkou je abirateróniumacetát. Jedna tableta obsahuje 250 mg abiraterónacetátu.
- Ďalšie zložky sú mikrokryštalická celulóza, sodná soľ kroskarmelózy, monohydrát laktózy; stearát horečnatý, povidón (K29 / K32), bezvodý koloidný oxid kremičitý a laurylsulfát sodný (pozri časť 2 „ZYTIGA obsahuje laktózu a sodík“).
Opis toho, ako ZYTIGA vyzerá a obsah balenia
- Tablety ZYTIGA sú oválneho tvaru, bielej až sivobielej farby, s vyrazeným „AA250“ na jednej strane.
- Tablety sa dodávajú v plastovej fľaši s plastovým uzáverom bezpečným pred deťmi. Každá fľaša obsahuje 120 tabliet. Každý box obsahuje jednu fľašu.
Zdrojový leták: AIFA (Talianska agentúra pre lieky). Obsah zverejnený v januári 2016. Súčasné informácie nemusia byť aktuálne.
Aby ste mali prístup k najaktuálnejšej verzii, odporúča sa navštíviť webovú stránku AIFA (Talianska agentúra pre lieky). Vylúčenie zodpovednosti a užitočné informácie.
01.0 NÁZOV LIEKU -
TABLETY ZYTIGA 250 MG
02.0 KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE -
Jedna tableta obsahuje 250 mg abiraterónacetátu.
Pomocné látky so známymi účinkami
Jedna tableta obsahuje 189 mg laktózy a 6,8 mg sodíka.
Úplný zoznam pomocných látok, pozri časť 6.1.
03.0 LIEKOVÁ FORMA -
Tablet
Biele až sivobiele tablety oválneho tvaru (15,9 mm dlhé x 9,5 mm široké) s vyrazeným AA250 na jednej strane.
04.0 KLINICKÉ INFORMÁCIE -
04.1 Terapeutické indikácie -
ZYTIGA je indikovaná spolu s prednizónom alebo prednizolónom na:
• liečba metastatického kastračne rezistentného karcinómu prostaty u asymptomatických alebo mierne symptomatických dospelých mužov po zlyhaní terapie depriváciou androgénov a u ktorých chemoterapia ešte nie je klinicky indikovaná (pozri časť 5.1).
• liečba metastatického kastračne rezistentného karcinómu prostaty u dospelých mužov, ktorých choroba pokročila počas režimu chemoterapie na báze docetaxelu alebo po ňom.
04.2 Dávkovanie a spôsob podávania -
Tento liek by mal predpisovať lekár so skúsenosťami s používaním protinádorových terapií.
Dávkovanie
Odporúčaná dávka je 1 000 mg (štyri 250 mg tablety), ktoré sa užívajú na prázdny žalúdok v jednej dennej dávke (pozri „Spôsob podávania“ nižšie). Užívanie tabliet s jedlom má za následok zvýšenie systémovej expozície abiraterónu (pozri časti 4.5 a 5.2).
ZYTIGA sa má užívať s nízkou dávkou prednizónu alebo prednizolónu. Odporúčaná dávka prednizónu alebo prednizolónu je 10 mg denne.
Lekárska kastrácia s analógom faktora uvoľňujúceho gonadotropín (hormón uvoľňujúci luteinizačný hormón, LHRH) by malo pokračovať počas liečby u pacientov, ktorí nie sú chirurgicky kastrovaní.
Pred začatím liečby sa majú zmerať hladiny sérových transamináz každé dva týždne počas prvých troch mesiacov liečby a potom každý mesiac. Monitorujte krvný tlak, zadržiavanie draslíka v sére a zadržiavanie tekutín každý mesiac (pozri časť 4.4). Pacienti so značným rizikom kongestívneho srdcového zlyhania by však mali byť sledovaní každé 2 týždne počas prvých troch mesiacov liečby a potom mesačne (pozri časť 4.4).
Uvažujte o udržaní hladín draslíka ≥ 4,0 mM u pacientov s už existujúcou hypokaliémiou alebo u tých, u ktorých sa počas liečby ZYTIGOU rozvinie hypokaliémia.
U pacientov, u ktorých sa vyvinie toxicita stupňa ≥ 3 vrátane hypertenzie, hypokaliémie, edému a iných nemineralokortikoidných toxicít, sa má liečba prerušiť a začať vhodná terapia. Liečba ZYTIGOU by nemala pokračovať, kým sa symptómy toxicity nezníži na 1. stupeň alebo na východiskový stav.
Ak sa vynechá denná dávka ZYTIGY, prednizónu alebo prednizolónu, v liečbe sa má pokračovať nasledujúci deň zvyčajnou dennou dávkou.
Hepatotoxicita
U pacientov, u ktorých sa počas liečby vyvinie hepatotoxicita (zvýšenie alanínaminotransferázy [ALT] alebo aspartátaminotransferázy [AST] o viac ako 5 -násobok hornej hranice normálu [ULN]), sa má liečba ihneď ukončiť (pozri časť 4.4). Obnovenie liečby, keď sa hodnoty pečeňových testov pacienta vrátia na východiskové hodnoty, možno vykonať zníženou dávkou 500 mg (dve tablety) jedenkrát denne. U pacientov, ktorí podstupujú opakovanú liečbu, sa majú hladiny sérových transamináz monitorovať najmenej každé dva týždne počas troch mesiacov a potom každý mesiac. Ak sa hepatotoxicita opakuje pri zníženej dávke 500 mg denne, liečba sa musí ukončiť.
Ak sa u pacientov kedykoľvek v priebehu terapie vyvinie ťažká hepatotoxicita (ALT alebo AST 20-násobok ULN), liečba sa má prerušiť a pacienti sa nemajú liečiť znova.
Poškodenie funkcie pečene
U pacientov s už existujúcou miernou poruchou funkcie pečene, Childova-Pughova trieda A, nie je potrebná žiadna úprava dávky.
Stredná porucha funkcie pečene (trieda B Child-Pughovej klasifikácie) má za následok približne štvornásobné zvýšenie systémovej expozície abiraterónu po jednorazových perorálnych dávkach abiraterónacetátu 1 000 mg (pozri časť 5.2). Nie sú k dispozícii žiadne klinické a bezpečnostné údaje. Účinnosť viacnásobných dávok abiraterón acetátu, ak sa podáva pacientom so stredne ťažkou alebo ťažkou poruchou funkcie pečene (Child-Plugh trieda B alebo C). Úpravu dávky nemožno očakávať. Použitie ZYTIGY je potrebné starostlivo zvážiť u pacientov so stredne ťažkou poruchou funkcie pečene, u ktorých musí prínos jasne prevyšovať možné riziko (pozri časti 4.2 a 5.2). ZYTIGA sa nemá používať u pacientov s ťažkou poruchou funkcie pečene (pozri časti 4.3, 4.4 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky (pozri časť 5.2). U pacientov s rakovinou prostaty a závažným poškodením funkcie obličiek nie sú žiadne klinické skúsenosti. U týchto pacientov sa odporúča opatrnosť (pozri časť 4.4).
Pediatrická populácia
Neexistuje žiadna indikácia pre špecifické použitie ZYTIGY v pediatrickej populácii.
Spôsob podávania
ZYTIGA je na perorálne použitie.
Tablety sa majú užívať najmenej dve hodiny po jedle a najmenej jednu hodinu po užití tabliet sa nemôže jesť. Tablety sa majú prehltnúť celé a zapiť malým množstvom vody.
04.3 Kontraindikácie -
- Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
- Tehotné ženy alebo ženy vo fertilnom veku (pozri časť 4.6).
• Závažná porucha funkcie pečene [stupnica C Childovej-Plughovej triedy C (pozri časti 4.2, 4.4 a 5.2)].
04.4 Špeciálne upozornenia a vhodné opatrenia pri používaní -
Hypertenzia, hypokaliémia, zadržiavanie tekutín a srdcové zlyhanie spôsobené nadbytkom mineralokortikoidov
ZYTIGA môže spôsobiť hypertenziu, hypokaliémiu a zadržiavanie tekutín (pozri časť 4.8), ako dôsledok zvýšených hladín mineralokortikoidov spôsobených inhibíciou CYP17 (pozri časť 5.1). Súbežné podávanie kortikosteroidov inhibuje aktivitu adrenokortikotropného hormónu (ACTH), čo má za následok zníženie výskytu a závažnosti týchto nežiaducich reakcií. Opatrnosť sa odporúča pri liečbe pacientov so základnými klinickými stavmi, ktoré môžu byť oslabené zvýšením krvného tlaku. z hypokaliémie (napr. pacienti liečení srdcovými glykozidmi) alebo zadržiavania tekutín (napr. pacienti so srdcovým zlyhaním) so závažnou alebo nestabilnou angínou pektoris, nedávnym infarktom myokardu alebo ventrikulárnou arytmiou a pacienti s poruchou funkcie obličiek striktne.
ZYTIGA sa má používať opatrne u pacientov s kardiovaskulárnym ochorením v anamnéze. Klinické štúdie fázy 3 vylúčili pacientov s nekontrolovanou hypertenziou, klinicky významnou srdcovou chorobou preukázanou infarktom myokardu alebo aterotrombotickými príhodami za posledných 6 mesiacov, závažnou alebo nestabilnou angínou alebo srdcovým zlyhaním triedy III alebo IV New York Heart Association (NYHA) (štúdia 301) alebo srdcové zlyhanie triedy II - IV (štúdia 302) alebo meranie srdcovej ejekčnej frakcie predsieňová fibrilácia alebo iné srdcové arytmie vyžadujúce lekársku terapiu. Bezpečnosť u pacientov s ejekčnou frakciou ľavej komory (LVEF)
Pred liečbou pacientov so značným rizikom kongestívneho srdcového zlyhania (napr. Srdcové zlyhanie v anamnéze, nekontrolovaná hypertenzia alebo srdcové príhody, ako je ischemická choroba srdca) zvážte vyšetrenie srdcových funkcií (napr. Echokardiogram). Pred liečbou ZYTIGOU sa má liečiť srdce zlyhanie a optimalizované srdcové funkcie. Hypertenziu, hypokaliémiu a zadržiavanie tekutín je potrebné korigovať a kontrolovať. Počas liečby je potrebné každé obdobie monitorovať krvný tlak, retenciu draslíka a tekutín v sére (prírastok hmotnosti, periférny edém) a akékoľvek ďalšie príznaky a príznaky kongestívneho srdcového zlyhania. 2 týždne po dobu 3 mesiacov a potom mesačne a opravené na abnormality. U pacientov s hypokaliémiou sa v súvislosti s liečbou ZYTIGOU pozorovalo predĺženie QT intervalu. Vyhodnoťte srdcovú funkciu, ako je klinicky indikované, začnite primeranú liečbu a zvážte prerušenie tejto liečby v prípade významného zníženia srdcových funkcií (pozri časť 4.2).
Hepatotoxicita a poškodenie funkcie pečene
V kontrolovaných klinických skúšaniach sa pozorovalo výrazné zvýšenie pečeňových enzýmov, ktoré viedlo k prerušeniu liečby alebo úprave dávky (pozri časť 4.8). Sérové hladiny transamináz sa majú merať každé dva týždne pred začiatkom liečby. Počas prvých troch mesiacov liečby a každý mesiac Ak sa vyvinú klinické príznaky a symptómy naznačujúce hepatotoxicitu, je potrebné okamžite zmerať sérové transaminázy. Ak sa v ktoromkoľvek čase ALT alebo AST zvýši 5-násobok ULN, liečba sa má ihneď ukončiť a funkcia pečene sa má starostlivo sledovať. . V liečbe je možné pokračovať zníženou dávkou až potom, čo sa hodnoty pečeňových testov pacienta vrátia na východiskové hodnoty (pozri časť 4.2).
Ak sa u pacientov vyvinie závažná hepatotoxicita (zvýšenie ALT alebo AST o 20-násobok HHN) kedykoľvek počas liečby, liečba sa musí prerušiť a takíto pacienti sa už nemajú liečiť.
Pacienti s aktívnou alebo symptomatickou vírusovou hepatitídou boli vylúčení z klinických štúdií; preto neexistujú žiadne údaje, ktoré by podporovali používanie ZYTIGY v tejto populácii.
Nie sú k dispozícii žiadne údaje o klinickej bezpečnosti a účinnosti viacnásobných dávok abiraterón acetátu pri podávaní pacientom so stredne ťažkou alebo ťažkou poruchou funkcie pečene (Child-Plugh trieda B alebo C). Použitie ZYTIGY sa má u pacientov hodnotiť opatrne. stredne ťažkou poruchou funkcie pečene, u ktorej musí prínos jasne prevyšovať možné riziko (pozri časti 4.2 a 5.2). ZYTIGA sa nesmie používať u pacientov s ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 5.2).
Po uvedení lieku na trh boli zriedkavo hlásené akútne zlyhanie pečene a fulminantná hepatitída, niektoré s fatálnym koncom (pozri časť 4.8).
Prerušenie podávania kortikosteroidov a liečba stresových situácií
Ak pacienti prerušia liečbu prednizónom alebo prednizolónom, odporúča sa opatrnosť a monitorovanie adrenokortikálnej insuficiencie. Ak ZYTIGA pokračuje aj po vysadení kortikosteroidov, pacienti majú byť sledovaní kvôli príznakom prebytku mineralokortikoidov (pozri informácie vyššie).
U pacientov na prednizóne alebo prednizolóne, ktorí sú vystavení neobvyklému stresu, možno pred stresovou situáciou, počas nej a po nej odporučiť zvýšenie dávky kortikosteroidov.
Hustota kostí
U mužov s metastatickým pokročilým karcinómom prostaty (kastračne rezistentný karcinóm prostaty) môže dôjsť k zníženiu hustoty kostí. Použitie ZYTIGY v kombinácii s glukokortikoidom môže tento účinok zosilniť.
Predchádzajúce použitie ketokonazolu
Pacienti s rakovinou prostaty, ktorí boli predtým liečení ketokonazolom, môžu dosiahnuť nižšiu mieru odpovede.
Hyperglykémia
Použitie glukokortikoidov môže zvýšiť hyperglykémiu, preto by sa u pacientov s cukrovkou mala často merať hladina glukózy v krvi.
Použitie v chemoterapii
Bezpečnosť a účinnosť ZYTIGY používanej súbežne s cytotoxickou chemoterapiou nebola stanovená (pozri časť 5.1).
Neznášanlivosť na pomocné látky
Tento liek obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami intolerancie galaktózy, lapónskeho deficitu laktázy alebo glukózo-galaktózovej malabsorpcie by nemali užívať tento liek. Tento liek navyše obsahuje viac ako 1 mmol (alebo 27,2 mg) sodíka v dávke štyroch tabliet. To je potrebné vziať do úvahy u pacientov na diéte so zníženým obsahom sodíka.
Potenciálne riziká
U mužov s metastatickým kastračne rezistentným karcinómom prostaty vrátane tých, ktorí sú liečení ZYTIGOU, sa môže vyskytnúť anémia a sexuálna dysfunkcia.
Účinky na kostrové svaly
U pacientov liečených ZYTIGOU boli hlásené prípady myopatie. Niektorí pacienti mali rabdomyolýzu s poruchou funkcie obličiek. Väčšina prípadov sa vyvinula počas prvého mesiaca liečby a ustúpila po prerušení podávania ZYTIGY. Opatrnosť sa odporúča u pacientov súbežne liečených liekmi, o ktorých je známe, že sú spojené s myopatiou / rabdomyolýzou.
Interakcie s inými liekmi
Vzhľadom na riziko zníženej expozície abiraterónu sa treba počas liečby vyhýbať silným induktorom CYP3A4, pokiaľ neexistuje terapeutická alternatíva (pozri časť 4.5).
04.5 Interakcie s inými liekmi a iné formy interakcie -
Vplyv jedla na abiraterón acetát
Podávanie s jedlom významne zvyšuje absorpciu abiraterón acetátu. Účinnosť a bezpečnosť pri podávaní s jedlom ešte nebola stanovená, preto sa tento liek nemá užívať s jedlom (pozri časti 4.2 a 5.2).
Interakcia s inými liekmi
Potenciál iných liekov ovplyvniť expozíciu abiraterónu
V klinickej farmakokinetickej interakčnej štúdii u zdravých jedincov vopred ošetrených silným induktorom CYP3A4 rifampicínom 600 mg denne počas 6 dní, po ktorom nasledovala jedna dávka abiraterón acetátu 1 000 mg, bola priemerná plazmatická AUC abiraterónu znížená o 55 %.
Silné induktory CYP3A4 (napr. Fenytoín, karbamazepín, rifampicín, rifabutín, rifapentín, fenobarbital, ľubovník bodkovaný [Hypericum perforatum]) sa majú počas liečby vyhýbať, pokiaľ neexistuje terapeutická alternatíva.
V ďalšej klinickej farmakokinetickej interakčnej štúdii u zdravých jedincov nemalo súbežné podávanie ketokonazolu, silného inhibítora CYP3A4, žiadny klinicky významný vplyv na farmakokinetiku abiraterónu.
Potenciál ovplyvniť expozíciu iných liekov
Abirateron je inhibítorom pečeňových enzýmov CYP2D6 a CYP2C8.
V štúdii na stanovenie účinkov abiraterón acetátu (plus prednizónu) s jednorazovou dávkou substrátu CYP2D6 dextrometorfánu sa systémová expozícia (AUC) dextrometorfánu zvýšila približne 2,9-násobne. AUC24 na dextrorfan, aktívny metabolit dextrometorfánu, sa zvýšil približne o 33%.
Pri podávaní liekov aktivovaných alebo metabolizovaných CYP2D6, obzvlášť liekov s nízkym terapeutickým indexom, sa odporúča opatrnosť. Má sa zvážiť zníženie dávky liekov s nízkym terapeutickým indexom metabolizovaných CYP2D6. Medzi príklady liekov metabolizovaných CYP2D6 patrí metoprolol, propranolol, desipramín, venlafaxín, haloperidol, risperidón, propafenón, flecanid, kodeín, oxykodón a tramadol (posledné tri lieky vyžadujú aktivitu CYP2D6 na tvorbu svojich aktívnych analgetických metabolitov).
V klinickej štúdii liekových interakcií CYP2C8 u zdravých jedincov sa AUC pioglitazónu zvýšila o 46% a AUC pre M-III a M-IV, aktívne metabolity pioglitazónu, sa pri podávaní pioglitazónu znížila o 10%. spolu s jednorazovou dávkou abirateróniumacetátu 1 000 mg. Aj keď tieto výsledky naznačujú, že pri kombinácii lieku ZYTIGA s liekmi, ktoré sú primárne vylučované CYP2C8, sa neočakávajú klinicky významné zvýšenia expozície, pacienti by mali byť starostlivo sledovaní z hľadiska prejavov toxicity súvisiacich s CYP2C8 pri súbežnom použití s substrátmi s úzkym terapeutickým indexom.
In vitroUkázalo sa, že hlavné metabolity abiraterónsulfát a N-oxid abiraterónsulfát inhibujú transportérpríjem pečeňovým OATP1B1 a v dôsledku toho to môže zvýšiť koncentrácie liekov eliminovaných OATP1B1. Nie sú k dispozícii žiadne klinické údaje na potvrdenie interakcie s transportérom.
Používajte s liekmi, o ktorých je známe, že predlžujú QT interval
Pretože terapia depriváciou androgénov môže predĺžiť QT interval, je potrebná opatrnosť pri podávaní ZYTIGY spolu s liekmi, o ktorých je známe, že predlžujú QT interval alebo s liekmi schopnými vyvolať torsade de pointes, ako sú antiarytmiká triedy IA (napr. Chinidín, disopyramid) alebo triedy III (napr. Amiodarón, sotalol, dofetilid, ibutilid), metadón, moxifloxacín, antipsychotiká atď.
Používajte so spironolaktónom
Spironolaktón viaže androgénny receptor a môže zvýšiť hladiny prostatického špecifického antigénu (PSA). Použitie so ZYTIGOU sa neodporúča (pozri časť 5.1).
04.6 Tehotenstvo a dojčenie -
Ženy vo fertilnom veku
Nie sú k dispozícii žiadne údaje o použití ZYTIGY u gravidných žien a použitie tohto lieku je kontraindikované u žien vo fertilnom veku.
Antikoncepcia u mužov a žien
Nie je známe, či sa abiraterón alebo jeho metabolity vylučujú do spermy. Ak má pacient počas tehotenstva pohlavný styk so ženou, odporúča sa použiť kondóm. Ak má pacient pohlavný styk so ženou vo fertilnom veku, odporúča sa použiť kondóm spolu s iným účinným antikoncepčným prostriedkom. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
Tehotenstvo
ZYTIGA nie je indikovaná u žien a je kontraindikovaná počas gravidity alebo u žien vo fertilnom veku (pozri časti 4.3 a 5.3).
Čas kŕmenia
Použitie ZYTIGY je u žien kontraindikované.
Plodnosť
Abiraterón ovplyvňuje plodnosť samcov a samíc potkanov, ale tieto účinky sú úplne reverzibilné (pozri časť 5.3).
04.7 Účinky na schopnosť viesť vozidlá a obsluhovať stroje -
ZYTIGA nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
04.8 Nežiaduce účinky -
Zhrnutie bezpečnostného profilu
Najčastejšie pozorovanými nežiaducimi reakciami sú periférny edém, hypokaliémia, hypertenzia a infekcie močových ciest.
Medzi ďalšie dôležité nežiaduce reakcie patrí ochorenie srdca, hepatotoxicita, zlomeniny a alergická alveolitída.
ZYTIGA môže spôsobiť hypertenziu, hypokaliémiu a zadržiavanie tekutín ako farmakodynamický dôsledok mechanizmu účinku. V klinických štúdiách boli očakávané nežiaduce reakcie mineralokortikoidov pozorované častejšie u pacientov liečených abiraterón acetátom ako u pacientov liečených placebom: hypokaliémia, 21. % vs 11%, hypertenzia 16% vs 11% a retencia tekutín (periférny edém) 26% vs 20%. U pacientov liečených abiraterón acetátom bola u 4% a 2% pacientov pozorovaná hypokaliémia 3. a 4. stupňa a hypertenzia 3. a 4. stupňa (spoločné terminologické kritériá pre nežiaduce udalosti, CTCAE, verzia 3.0). Reakcie mineralokortikoidov boli zvládnuté farmakologicky s pozitívnymi výsledkami. Súbežné používanie kortikosteroidov znižuje výskyt a závažnosť týchto nežiaducich reakcií (pozri časť 4.4).
Tabuľka nežiaducich reakcií
Štúdie vykonané na pacientoch s pokročilým metastatickým karcinómom prostaty, ktorí dostávali analóg LHRH alebo ktorí v minulosti podstúpili orchiektómiu, zahŕňali podanie dávky ZYTIGY 1 000 mg denne v kombinácii s nízkou dávkou prednizónu alebo prednizolónu (10 mg na deň).
Nežiaduce reakcie na liek pozorované počas klinických štúdií a skúseností po uvedení lieku na trh sú uvedené nižšie podľa kategórie frekvencie. Kategórie frekvencií sú definované nasledovne: veľmi časté (≥ 1/10); časté (≥ 1/100,
V rámci každej kategórie frekvencie sú nežiaduce účinky uvedené v poradí klesajúcej závažnosti. Tabuľka 1: Nežiaduce reakcie identifikované v klinických a postmarketingových štúdiách
* Srdcové zlyhanie zahŕňa aj kongestívne srdcové zlyhanie, zlyhanie ľavej komory a zníženú ejekčnú frakciu.
** Zlomeniny zahŕňajú všetky zlomeniny okrem patologickej zlomeniny
a Spontánne správy z postmarketingových skúseností
U pacientov liečených abiraterón acetátom sa vyskytli nasledujúce nežiaduce reakcie 3. stupňa (CTCAE verzia 3.0): hypokaliémia 3%; infekcia močových ciest, zvýšenie alanínaminostransferázy, hypertenzia, zvýšenie aspartátaminotransferázy, zlomeniny 2%; periférny edém, srdcové zlyhanie a fibrilácia predsiení po 1%. Hypertriglyceridémia a angina pectoris 3. stupňa (CTCAE verzia 3.0) sa vyskytli v
Popis vybraných nežiaducich reakcií
Kardiovaskulárne reakcie
Obe klinické štúdie fázy 3 vylúčili pacientov s nekontrolovanou hypertenziou, klinicky významným srdcovým ochorením, ktoré boli dokázané infarktom myokardu alebo aterotrombotickými príhodami za posledných 6 mesiacov, závažnou alebo nestabilnou angínou alebo srdcovým zlyhaním triedy NYHA III alebo IV (štúdia 301) alebo srdcovým zlyhaním trieda II - IV (štúdia 302) o meranie apoplexie srdcovej ejekčnej frakcie a náhlej srdcovej smrti. V klinických štúdiách fázy 3 bol výskyt nežiaducich reakcií vaskulárneho typu u pacientov užívajúcich abiraterón acetát oproti pacientom užívajúcim placebo: hypertenzia 14,5% proti 10,5%, fibrilácia predsiení 3,4% proti 3,4%, tachykardia 2,8% proti 1,7%, angina pectoris 1,9% proti 0,9%, srdcové zlyhanie 1,9% proti 0,6%a arytmia 1,1% proti 0,4%.
Hepatotoxicita
U pacientov liečených abiraterón acetátom bola hlásená hepatotoxicita so zvýšením ALT, AST a celkového bilirubínu.Vo všetkých klinických štúdiách bolo zvýšenie pečeňových funkčných testov (zvýšenie ALT alebo AST> 5 x ULN [horná hranica normy] alebo bilirubínu> 1,5 x ULN) hlásené u približne 4% pacientov, ktorí dostávali abiraterón acetát, zvyčajne počas prvé 3 mesiace od začiatku liečby. V klinickej štúdii 301 mali pacienti so zvýšenou východiskovou hodnotou ALT alebo AST vyššie hodnoty pečeňových funkčných testov ako pacienti, ktorí začali s normálnymi hodnotami. Pri zvýšenej hladine ALT alebo AST> 5 x ULN alebo zvýšení bilirubínu> Boli pozorované 3 x ULN, abiraterón acetát bol prerušený alebo prerušený. V dvoch prípadoch došlo k výraznému zvýšeniu pečeňových funkčných testov (pozri časť 4.4) Dvaja pacienti s normálnou funkciou pečene na začiatku mali zvýšenia ALT alebo AST od 15 do 40 x ULN a v bilirubíne od 2 do 6 x ULN. testy pečeňových funkcií u oboch pacientov sa vrátili do normálu a jeden pacient prešiel opakovanou liečbou bez opakovaného zvýšenia hodnôt. V štúdii 302 bolo pozorované zvýšenie ALT alebo AST 3. alebo 4. stupňa u 35 pacientov (6,5%) liečených abiraterón acetátom. Zvýšenie aminotransferázy ustúpilo u všetkých okrem 3 pacientov (2 s novými viacnásobnými pečeňovými metastázami a 1 so zvýšením AST približne 3 týždne po poslednej dávke abiraterón acetátu). Prerušenie liečby kvôli zvýšeniu ALT a AST bolo hlásené u 1,7% a 1,3% pacientov liečení abiraterón acetátom a 0,2% respektíve 0% pacientov liečených placebom; v dôsledku hepatotoxických príhod nebolo hlásené žiadne úmrtie.
V klinických skúšaniach bolo riziko hepatotoxicity zmiernené vylúčením pacientov s východiskovou hepatitídou alebo s významnými abnormalitami pečeňových testov. V klinickom skúšaní 301 boli pacienti s východiskovými hodnotami ALT a AST ≥ 2,5 -násobok ULN, bez pečeňových metastáz a> 5 x ULN, pečeňové metastázy boli vylúčené. V klinickej štúdii nebolo zaradených 302 pacientov s pečeňovými metastázami a boli vylúčení pacienti s východiskovými hodnotami ALT a AST ≥ 2,5 x ULN. Abnormality testov funkcie pečene, pozorované u pacientov, ktorí sa zúčastnili klinických skúšaní, boli zvládnuté dynamicky tak, že sa uchýlia k prerušeniu terapie a umožnia opakovanie liečby až potom, ako sa hodnoty v testoch pečeňových funkcií vrátia na východiskové hladiny pacienta (pozri časť 4.2). Pacienti so zvýšením ALT alebo AST> 20-násobok ULN nepodstúpili opakovanú liečbu. Bezpečnosť opakovanej liečby u týchto pacientov nie je známa. Mechanizmus hepatotoxicity spojenej so ZYTIGOU nie je známy.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité, pretože umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia. "Adresa www. agenziafarmaco.gov.it/it/responsabili.
04.9 Predávkovanie -
Skúsenosti s predávkovaním ZYTIGOU u ľudí sú obmedzené.
Neexistuje žiadne špecifické antidotum. V prípade predávkovania sa má dávkovanie prerušiť a vykonať všeobecné podporné opatrenia vrátane monitorovania arytmií, hypokaliémie a prejavov a symptómov zadržiavania tekutín. Má sa vykonať aj vyhodnotenie funkcie pečene.
05.0 FARMAKOLOGICKÉ VLASTNOSTI -
05.1 "Farmakodynamické vlastnosti -
Farmakoterapeutická skupina: endokrinná terapia, iné hormonálne antagonisty a príbuzné látky.
ATC kód: L02BX03.
Mechanizmus akcie
Abiraterón acetát (ZYTIGA) sa prevedie in vivo v abiraterone, inhibítore biosyntézy androgénov. Abiraterón konkrétne selektívne inhibuje enzým 17α-hydroxylázu / C17,20-lyázu (CYP17). Tento enzým je normálne exprimovaný a je potrebný pre biosyntézu androgénnych hormónov v semenníkových, nadobličkových a neoplastických tkanivách prostaty. CYP17 katalyzuje konverziu pregnenolónu a progesterónu na prekurzory testosterónu, DHEA a androstendión, 17α-hydroxyláciou a štiepením väzby C17,20. Inhibícia CYP17 tiež spôsobuje zvýšenie produkcie mineralokortikoidov v nadobličkách (pozri časť 4.4).
Androgén-senzitívna rakovina prostaty reaguje na liečbu znížením hladiny androgénu. Terapie deprivácie androgénu, ako je liečba analógmi LHRH alebo orchiektómia, znižujú produkciu androgénov v semenníkoch bez toho, aby mali akýkoľvek vplyv na produkciu androgénnych hormónov v nadobličkách alebo v nádore. Liečba ZYTIGOU znižuje hladinu testosterónu v sére na nedetekovateľné hladiny (pomocou komerčných testov), ak sa podáva s analógmi LHRH (alebo po orchiektómii).
Farmakodynamické účinky
ZYTIGA znižuje hladinu testosterónu v sére a ďalších androgénnych hormónov na nižšie hodnoty, než aké sa dosahujú použitím analógov LHRH alebo samotnej orchiektómie. Tento účinok je dôsledkom selektívnej inhibície enzýmu CYP17 potrebného na biosyntézu androgénov. PSA funguje ako biomarker v pacienti s rakovinou prostaty. V klinickej štúdii fázy 3, ktorá sa uskutočnila u pacientov s progresiou po predchádzajúcej chemoterapii taxánmi, 38% pacientov liečených abiraterón acetátom preukázalo zníženie najmenej o 50% hladín PSA oproti východiskovým hodnotám v porovnaní s 10% pacientov, ktorí dostali placebo.
Klinická účinnosť a bezpečnosť
Účinnosť bola stanovená v dvoch multicentrických, randomizovaných, placebom kontrolovaných klinických štúdiách fázy 3 (štúdie 301 a 302) u pacientov s metastatickým kastračne rezistentným karcinómom prostaty. Do štúdie 302 boli zaradení pacienti, ktorí predtým neboli liečení docetaxelom, a do štúdie 301 zaradených pacientov, ktorí mali predtým pacienti dostávali analóg LHRH alebo predtým podstúpili orchiektómiu. V ramene s liečbou účinnou látkou sa ZYTIGA podávala v dávke 1 000 mg denne v kombinácii s nízkou dávkou prednizónu alebo prednizolónu 5 mg dvakrát denne. Pacienti v kontrolnej skupine dostávali placebo a nízku dávku prednizónu alebo prednizolónu 5 mg dvakrát denne.
Variácie zistené v sérovej koncentrácii PSA oddelene nie vždy predpovedajú klinický prínos. Preto v oboch klinických skúšaniach bolo odporúčané, aby pacienti udržiavali liečebný režim s priradenou študijnou liečbou, kým nebudú pre každú klinickú štúdiu splnené nižšie uvedené kritériá prerušenia.
V oboch štúdiách nebolo používanie spironolaktónu povolené, pretože viaže androgénny receptor a môže zvýšiť hladiny PSA.
Štúdia 302 (pacienti bez predchádzajúcej chemoterapie)
Do tejto štúdie boli zaradení pacienti bez predchádzajúcej chemoterapie, ktorí boli asymptomatickí alebo mierne symptomatickí a u ktorých chemoterapia ešte nebola klinicky indikovaná. Intenzívnejšia epizóda bolesti za posledných 24 hodín so skóre 0-1 bola považovaná za asymptomatickú sekundu Stručná inventúra bolesti-krátka forma (BPI-SF) a skóre 2-3 boli považované za mierne symptomatické.
V štúdii 302 (n = 1088) bol priemerný vek zaradených pacientov 71 rokov pre pacientov liečených ZYTIGOU plus prednizón alebo prednizolón a 70 rokov pre pacientov liečených placebom plus prednizón alebo prednizolón. Počet pacientov liečených ZYTIGOU podľa rasy skupina bola 520 belochov (95,4%), 15 čiernych (2,8%), 4 ázijské (0,7%) a 6 ďalších (1,1%).Východná družstevná onkologická skupina (ECOG) bol 0 pre 76% pacientov a 1 pre 24% pacientov v oboch ramenách. Päťdesiat percent pacientov malo iba kostné metastázy, ďalších 31% pacientov malo metastázy v kostiach a mäkkých tkanivách alebo lymfatických uzlinách a 19% pacientov malo iba metastázy v mäkkých tkanivách alebo lymfatických uzlinách. Pacienti s viscerálnymi metastázami boli vylúčení. The koncový bod primárne skóre účinnosti bolo celkové prežitie a prežitie bez rádiologickej progresie (rPFS). Okrem veľkosti koncový bod prínos sa tiež hodnotil s použitím času používania opioidov na liečbu rakoviny, času do začiatku cytotoxickej chemoterapie, času do regresie ≥ 1 bodového skóre ECOG a času do progresie PSA na základe kritérií Pracovná skupina pre rakovinu prostaty-2 (PCWG2). Študijné ošetrenia boli prerušené v čase jednoznačnej klinickej progresie. Liečba mohla byť tiež prerušená, podľa uváženia skúšajúceho, v čase potvrdenej rádiologickej progresie.
Prežitie bez rádiologickej progresie (rPFS) bolo hodnotené s použitím zobrazovanie sekvenčné, ako sú definované kritériami PCWG2 (pre kostné lézie) a upravenými kritériami Kritériá vyhodnotenia reakcie pri solídnych nádoroch (RECIST) (pri poraneniach mäkkých tkanív). Analýzy rPFS používali centrálne hodnotené rádiologické hodnotenie progresie.
Pri "plánovanej analýze rPFS c" bolo 401 udalostí, 150 (28%) pacientov liečených ZYTIGOU a 251 (46%) pacientov liečených placebom malo rádiologický dôkaz progresie alebo zomrelo. Medzi liečebnými skupinami bol pozorovaný významný rozdiel v rPFS (pozri tabuľku 2).
NE = Neodhaduje sa
* P-hodnota založená na log-rank teste upravená o stratifikačné faktory ECOG (0 alebo 1)
** Pomer rizika
Zhromažďovanie údajov o pacientoch však pokračovalo až do dátumu druhej analýzy medzitým celkové prežitie (celkové prežitie - OS). Rádiologické vyšetrenie rPFS vyšetrovateľom je uvedené v tabuľkách 3.
Šesťsto sedem pacientov malo rádiologickú progresiu alebo zomrelo: 271 (50%) v skupine s abiraterón acetátom a 336 (62%) v skupine s placebom. Liečba abiraterón acetátom znížila riziko rádiologickej progresie alebo smrti o 47% v porovnaní s placebom (HR = 0,530; 95% CI: [0,451; 0,623], s
* P-hodnota založená na log-rank teste upravená o stratifikačné faktory ECOG (0 alebo 1)
** Pomer rizika
Plánovaná predbežná analýza (IA) pre OS bola vykonaná po pozorovaní 333 úmrtí. Na základe pozorovaného významného klinického prínosu bola štúdia otvorená a liečba liekom ZYTIGA bola ponúkaná pacientom v skupine s placebom. Prežitie bolo celkovo pre ZYTIGA dlhšie ako pre placebo s 25% zníženie rizika úmrtia (HR = 0,752; 95% CI: [0,606; 0,934], p = 0,0097), ale OS nebol vyspelý a výsledky ad interim nespĺňali cieľové zastavovacie limity pre štatistickú významnosť (pozri Tabuľka 4). Po tejto AI pokračovalo prežitie.
Konečná plánovaná analýza OS bola vykonaná po pozorovaní 741 úmrtí (medián sledovania 49 mesiacov). Šesťdesiatpäť percent pacientov (354 z 546) liečených ZYTIGOU v porovnaní so 71% (387 z 542).) Pacientov liečení placebom, zomreli. Štatisticky významná výhoda OS v skupine ZYTIGA bola preukázaná s 19,4% znížením rizika smrti (HR = 0,806; 95% CI: [0,697; 0,931], p = 0,0033) a medián zlepšenia OS 4,4 mesiaca (ZYTIGA 34,7 mesiaca, placebo 30,3 mesiaca) (pozri tabuľku 4). Toto zlepšenie bolo preukázané napriek tomu, že 44% pacientov v placebe dostalo ZYTIGA ako následnú liečbu.
NE = Neodhaduje sa
* P-hodnota založená na log-rank teste upravená o stratifikačné faktory ECOG (0 alebo 1)
** Pomer rizika
Okrem pozorovaných zlepšení celkového prežívania a rPFS bol prínos preukázaný aj pri liečbe ZYTIGOU proti placebo vo všetkých koncový bod sekundárne nasledovne:
Čas do progresie PSA na základe kritérií PCWG2: Medián času do progresie PSA bol 11,1 mesiaca u pacientov, ktorí dostávali ZYTIGA, a 5,6 mesiaca u pacientov, ktorí dostávali placebo (HR = 0,488; 95% IS: [0,420; 0,568], s
Čas do užívania opioidov pri rakovinovej bolesti: Medián času do užívania opioidov pri bolesti spôsobenej rakovinou prostaty v čase konečnej analýzy bol 33,4 mesiaca u pacientov užívajúcich ZYTIGA a 23,4 mesiacov u pacientov, ktorí dostávali placebo (HR = 0,721; 95% CI: [0,614, 0,846], s
Čas do cytotoxickej chemoterapie: Medián času do cytotoxickej chemoterapie bol 25,2 mesiacov u pacientov, ktorí dostávali ZYTIGA, a 16,8 mesiaca u pacientov, ktorí dostávali placebo (HR = 0,580; 95% CI: [0,487; 0,691], s
Čas do zhoršenia skóre ECOG ≥ 1 bod: Medián času do zhoršenia skóre ECOG ≥ 1 bod bol 12,3 mesiaca u pacientov užívajúcich ZYTIGA a 10,9 mesiaca u pacientov užívajúcich placebo (HR = 0,821; 95% CI: [0,714, 0,943], p = 0,0053).
Nasledujúce ukazovatele preukázali štatisticky významnú výhodu v prospech liečby ZYTIGOU:
Objektívna odpoveď: Objektívna odpoveď bola definovaná ako percento pacientov s merateľným ochorením, ktorí dosiahli úplnú alebo čiastočnú odpoveď podľa kritérií RECIST (za cieľovú léziu sa požadovala východisková veľkosť lymfatických uzlín ≥ 2 cm). Percento pacientov s merateľným ochorením na začiatku s objektívnou odpoveďou bolo 36% v skupine so ZYTIGA a 16% v skupine s placebom (p
Bolesť: Liečba ZYTIGOU významne znížila riziko progresie priemernej intenzity bolesti o 18% v porovnaní so skupinou s placebom (p = 0,0490). Medián času do progresie bol 26,7 mesiaca v skupine so ZYTIGA a 18, 4 mesiacov v skupine s placebom.
Čas do zhoršenia FACT-P (celkové skóre): Liečba ZYTIGOU znížila riziko zhoršenia FACT-P (celkové skóre) o 22% v porovnaní s placebom (p = 0,0028). Medián času do zhoršenia FACT-P (celkové skóre) bol 12,7 mesiaca v skupine so ZYTIGOU a 8,3 mesiaca v skupine s placebom.
Štúdia 301 (pacienti, ktorí predtým absolvovali chemoterapiu)
Do štúdie 301 boli zaradení pacienti, ktorí predtým dostávali docetaxel. Počas docetaxelu sa nepožadovalo, aby pacienti postupovali, pretože toxicita voči tejto chemoterapii mohla viesť k jej prerušeniu. Pacienti pokračovali v študijnom ošetrení až do progresie PSA (potvrdené 25% zvýšenie od východiskových / nižších hladín pacienta) spolu s rádiologickou progresiou definovanou v protokole a symptomatickou alebo klinickou progresiou. Pacienti s predchádzajúcou liečbou rakoviny prostaty ketokonazolom boli z tejto štúdie vylúčení. L "koncový bod primárnou účinnosťou bolo celkové prežitie.
Priemerný vek zaradených pacientov bol 69 rokov (rozsah 39-95). Počet pacientov liečených ZYTIGOU podľa rasovej skupiny bol 737 belochov (93,2%), 28 čiernych (3,5%), 11 ázijských (1,4%) a 14 ďalších (1,8%). Skóre malo 11% zaradených pacientov skóre výkonu podľa stupnice ECOG 2; 70% predložilo rádiografický dôkaz progresie ochorenia s progresiou PSA alebo bez nej; 70% absolvovalo predchádzajúcu cytotoxickú chemoterapiu a 30% malo dve. Metastázy v pečeni boli prítomné u 11% pacientov liečených ZYTIGOU.
V plánovanej analýze uskutočnenej po 552 úmrtiach zomrelo 42% (333 zo 797) pacientov liečených ZYTIGOU v porovnaní s 55% (219 z 398) pacientov liečených placebom. Štatisticky významné zlepšenie placeba bolo pozorované medián celkového prežívania pacientov liečených ZYTIGOU (pozri tabuľku 5).
p-hodnota založená na log-rank teste upravenom o stratifikačné faktory ECOG (0-1 vs 2), skóre bolesti (chýba vs prítomné), počet predchádzajúcich režimov chemoterapie (1 vs 2) a typ progresie ochorenia (iba PSA verzus rádiologický).
b Pomer rizika založený na rizikových modeloch upravený o stratifikačné faktory. Pomer rizika
Vo všetkých fázach hodnotenia zostal po prvých niekoľkých mesiacoch liečby nažive vyšší podiel pacientov liečených ZYTIGOU ako podiel pacientov, ktorí dostávali placebo.
Analýzy podskupín prežitia ukázali významný prínos pre prežitie pri liečbe ZYTIGOU.
Okrem pozorovaného zlepšenia celkového prežitia všetky koncový bod Sekundári štúdie boli v prospech lieku ZYTIGA a boli tiež štatisticky významní po úprave pre viacero štúdií na základe týchto skutočností:
Pacienti liečení ZYTIGOU mali významne vyššiu celkovú mieru odpovede PSA (definovanú ako zníženie o ≥ 50% oproti východiskovému stavu) v porovnaní s pacientmi, ktorí dostávali placebo, 38% oproti 10%, p
Priemerný čas do progresie PSA bol 10,2 mesiaca u pacientov liečených ZYTIGOU a 6,6 mesiacov u pacientov liečených placebom (HR = 0,580; 95% CI: [0,462; 0,728], p
Priemerné prežívanie bez progresie, stanovené rádiologickým vyšetrením, bolo 5,6 mesiaca u pacientov liečených ZYTIGOU a 3,6 mesiaca u pacientov liečených placebom (HR = 0,673; 95% CI: [0,585; 0,776], s
Ache
Percento pacientov, ktorí uviedli úľavu od bolesti, bolo štatisticky významne vyššie v skupine s liekom ZYTIGA ako v skupine s placebom (44% vs 27%, p = 0,0002). odpovedač na úľavu od bolesti bol definovaný ako pacient, u ktorého došlo za posledných 24 hodín k zníženiu najhoršieho skóre intenzity bolesti podľa BPI SF o najmenej 30% oproti východiskovým hodnotám, pričom pri dvoch po sebe nasledujúcich hodnoteniach nedošlo k zvýšeniu skóre použitia analgetík s odstupom štyroch týždňov .. Na úľavu od bolesti boli analyzované (n = 512) iba pacienti so skóre ≥ 4 a najmenej jedným post-východiskovým skóre bolesti.
Menšie percento pacientov liečených ZYTIGOU malo progresiu bolesti ako pacienti, ktorí užívali placebo v 6 (22% vs 28%), 12 (30% vs 38%) a 18 mesiacoch (35% vs 46%). Progresia bolesti bola definovaná ako ≥ 30% nárast od východiskového skóre v skóre najhoršej intenzity bolesti BPI SF za posledných 24 hodín bez toho, že by sa pozorovalo zníženie skóre použitia analgetika pri dvoch po sebe nasledujúcich návštevách alebo o ≥ 30% zvýšenie skóre v použití analgetika pozorované pri dve po sebe idúce návštevy. Čas do progresie bolesti do 25. percentilu bol 7,4 mesiaca v skupine s liekom ZYTIGA v porovnaní so 4,7 mesiaca v skupine s placebom.
Udalosti ovplyvňujúce kostrový systém
Nižšie percento pacientov v skupine s liekom ZYTIGA zažilo udalosti kostrového systému v porovnaní s pacientmi v skupine s placebom po dobu 6 mesiacov (18% oproti 28%), 12 mesiacov (30% oproti 40%) a 18 mesiacov (35% oproti 40%) . V skupine liečenej ZYTIGOU bol čas do prvej skeletálnej príhody v 25. percentile dvakrát dlhší ako v kontrolnej skupine za 9,9 mesiaca oproti 4,9 mesiaca. Udalosť kostrového systému bola definovaná ako patologická zlomenina, kompresia miechy, paliatívne ožarovanie kosti alebo chirurgický zákrok na kosti.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so ZYTIGOU vo všetkých podskupinách pediatrickej populácie s pokročilým karcinómom prostaty. Informácie o použití v pediatrickej populácii nájdete v časti 4.2.
05.2 "Farmakokinetické vlastnosti -
Po podaní abiraterón acetátu bol farmakokinetický profil abiraterónu a abiraterón acetátu študovaný u zdravých osôb, u pacientov s pokročilým metastatickým karcinómom prostaty a u nerakovinových subjektov s poruchou funkcie pečene alebo obličiek. Abiraterón acetát sa rýchlo premieňa in vivo v abiraterone, inhibítore biosyntézy androgénov (pozri časť 5.1).
Absorpcia
Po perorálnom podaní abiraterón acetátu nalačno je čas potrebný na dosiahnutie maximálnej plazmatickej koncentrácie abiraterónu približne 2 hodiny.
Podanie abiraterón acetátu s jedlom v porovnaní s podaním nalačno má za následok zvýšenie priemernej systémovej expozície abiraterónu až o 10 -násobok [AUC] a až 17 -násobok [Cmax] vyšší, vzhľadom na tuk obsiahnutý v jedle Vzhľadom na normálne odchýlky v obsahu a zložení jedál môže užívanie ZYTIGY s jedlom viesť k veľmi variabilným expozíciám. ZYTIGA sa preto nemá užívať s jedlom. Má sa užiť najmenej jednu hodinu pred jedlom alebo najmenej dve hodiny po jedle.Tablety sa majú prehltnúť celé a zapiť trochou vody (pozri časť 4.2).
Distribúcia
Väzba 14C-značeného abiraterónu na plazmatické proteíny je 99,8%. Zdanlivý distribučný objem je približne 5 630 l, čo naznačuje rozsiahlu distribúciu abiraterónu v periférnych tkanivách.
Biotransformácia
Po podaní 14C rádioaktívneho izotopom značeného abiraterón acetátu v kapsulách sa abiraterón acetát hydrolyzuje na abiraterón, ktorý sa potom podrobí metabolizmu, vrátane sulfatácie, hydroxylácie a oxidácie, predovšetkým v pečeni. Väčšina rádioaktivity prítomnej v obehu (približne 92%) sa nachádzala vo forme metabolitov abiraterónu. Dva hlavné metabolity 15 detegovateľných, abiraterónsulfát a N-oxid abiraterónsulfát, každý predstavuje približne 43% z celkovej rádioaktivity.
Vylúčenie
Priemerný polčas abiraterónu v plazme je približne 15 hodín, na základe údajov od zdravých jedincov. Po perorálnom podaní dávky 1 000 mg abirateróniumacetátu značeného rádioaktívnym izotopom 14C sa približne 88% dávky rádioaktivity zachytilo v stolici a 5% circanelového "moču. Hlavnými zlúčeninami prítomnými vo výkaloch sú nezmenený abiraterón acetát a abiraterón (približne 55% a 22% podanej dávky).
Poškodenie funkcie pečene
Farmakokinetika abiraterón acetátu sa skúmala u subjektov s už existujúcou miernou alebo stredne závažnou poruchou funkcie pečene (Childova-Pughova trieda A a B v uvedenom poradí) a u zdravých kontrolných subjektov. Systémová expozícia abiraterónu po jednorazovej perorálnej dávke 1 000 mg sa zvýšila o približne 11%, respektíve 260%, u subjektov s už existujúcou miernou a stredne závažnou poruchou funkcie pečene. Priemerný polčas abiraterónu bol predĺžený na približne 18 hodín u subjektov s miernou poruchou funkcie pečene a na približne 19 hodín u osôb so stredne ťažkou poruchou funkcie pečene.
V ďalšej klinickej štúdii bola farmakokinetika abiraterónu skúmaná u subjektov s už existujúcou ťažkou poruchou funkcie pečene (n = 8) (Child-Pugh trieda C) a u 8 zdravých kontrolných subjektov s normálnou funkciou pečene. AUC abiraterónu sa zvýšila o približne 600% a voľná frakcia liečiva o 80% u osôb s ťažkým poškodením funkcie pečene v porovnaní s osobami s normálnou funkciou pečene.
U pacientov s už existujúcou miernou poruchou funkcie pečene nie je potrebná žiadna úprava dávky.
Použitie abirateróniumacetátu je potrebné starostlivo zvážiť u pacientov so stredne ťažkou poruchou funkcie pečene, u ktorých musí prínos jasne prevyšovať možné riziko (pozri časti 4.2 a 4.4). Abirateróniumacetát sa nemá používať u pacientov s ťažkou poruchou funkcie pečene (pozri časti 4.2) 4,3 a 4,4).
U pacientov, u ktorých sa počas liečby vyvinie hepatotoxicita, môže byť potrebné prerušenie liečby a úprava dávky (pozri časti 4.2 a 4.4)..
Porucha funkcie obličiek
Farmakokinetika abiraterón acetátu sa porovnávala u pacientov s terminálnym štádiom ochorenia obličiek, ktorí podstupujú stabilný režim hemodialýzy, s porovnateľnými kontrolnými subjektmi s normálnou funkciou obličiek. Systémová expozícia abiraterónu po jednorazovej perorálnej dávke 1 000 mg nebola zvýšená u pacientov v terminálnom štádiu ochorenia obličiek, ktorí podstupujú dialýzu. Podávanie pacientom s poruchou funkcie obličiek, vrátane závažnej, nevyžaduje zníženie dávky (pozri časť 4.2. Nie sú však žiadne klinické skúsenosti. u pacientov s rakovinou prostaty a závažným poškodením funkcie obličiek. U týchto pacientov sa odporúča opatrnosť.
05.3 Predklinické údaje o bezpečnosti -
Vo všetkých štúdiách toxicity na zvieratách bolo pozorované významné zníženie hladín cirkulujúceho testosterónu. Výsledkom bolo zníženie hmotnosti orgánu a morfologické a / alebo histopatologické zmeny v reprodukčných orgánoch a nadobličkách, hypofýze a mliečnych žľazách. Všetky zmeny vykazovali úplnú alebo čiastočnú reverzibilitu. Zmeny v reprodukčných orgánoch a tých, ktoré sú citlivé na androgénne hormóny, sú kompatibilné s farmakológiou abiraterónu. Všetky zmeny hormónov súvisiace s liekom sa zvrátili alebo ustúpili po 4-týždňovom období zotavenia.
V štúdiách fertility na samcoch aj samiciach potkanov abirateróniumacetát znižoval fertilitu, čo je účinok, ktorý je plne reverzibilný 4 až 16 týždňov po prerušení abiraterón acetátu.
V štúdii vývojovej toxicity na potkanoch ovplyvnil abiraterón acetát graviditu vrátane zníženia hmotnosti plodu a prežitia. Účinky na vonkajšie genitálie boli pozorované, hoci abiraterón acetát nebol teratogénny.
V týchto štúdiách fertility a vývojovej toxicity na potkanoch všetky účinky korelovali s farmakologickou aktivitou abiraterón acetátu.
Okrem variácií zistených v reprodukčných orgánoch vo všetkých toxikologických štúdiách vykonaných na zvieratách, neklinické údaje neodhalili žiadne osobitné riziko pre ľudí na základe konvenčných štúdií farmakológia bezpečnosti, toxicita po opakovaných dávkach, genotoxicita a karcinogénny potenciál. Abiraterón acetát nebol v 6-mesačnej štúdii na transgénnych myšiach (Tg.rasH2) karcinogénny. V 24-mesačnej štúdii karcinogenity na potkanoch zvýšil abiraterón acetát výskyt semenníkových novotvarov v semenníkoch. Predpokladá sa, že toto zistenie súvisí s farmakologickým účinkom abiraterónu a je špecifické pre potkany. Abiraterón acetát nebol u samíc potkanov karcinogénny.
Účinná látka abiraterón predstavuje riziko pre vodné prostredie, najmä pre ryby.
06.0 FARMACEUTICKÉ INFORMÁCIE -
06.1 Pomocné látky -
Mikrokryštalická celulóza
Sodná soľ kroskarmelózy
Monohydrát laktózy
Stearan horečnatý
Povidón (K29 / K32)
Koloidný oxid kremičitý bezvodý
Laurylsulfát sodný
06.2 Nekompatibilita “-
Nie je to relevantné.
06.3 Obdobie platnosti “-
2 roky.
06.4 Špeciálne opatrenia na uchovávanie -
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
06.5 Charakter vnútorného obalu a obsah balenia -
Okrúhle biele polyetylénové fľaše s vysokou hustotou s polypropylénovým uzáverom bezpečným pred deťmi obsahujúce 120 tabliet. Každé balenie obsahuje jednu fľašu.
06.6 Pokyny na použitie a zaobchádzanie s liekom -
Vzhľadom na mechanizmus účinku môže tento liek poškodiť vyvíjajúci sa plod; preto by tehotné ženy alebo ženy v plodnom veku nemali s ním zaobchádzať bez použitia ochranných prostriedkov, akými sú napríklad rukavice.
Nepoužitý liek a odpad z tohto lieku musí byť zlikvidovaný v súlade s miestnymi predpismi. Tento liek môže predstavovať riziko pre vodné prostredie (pozri časť 5.3).
07.0 DRŽITEĽ „REGISTRÁCIE NA REGISTRÁCII“ -
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 Beerse
Belgicko
08.0 REGISTRAČNÉ ČÍSLO -
EU/1/11/714/001
041427016
09.0 DÁTUM PRVEJ REGISTRÁCIE ALEBO OBNOVENIA REGISTRÁCIE -
Dátum prvej registrácie: 5. september 2011
Dátum posledného obnovenia: 26. mája 2016
10.0 DÁTUM REVÍZIE TEXTU -
11/2016