Všeobecnosť
Termín retinitis pigmentosa (RP) identifikuje skupinu genetických chorôb charakterizovaných progresívnou degeneráciou sietnice.

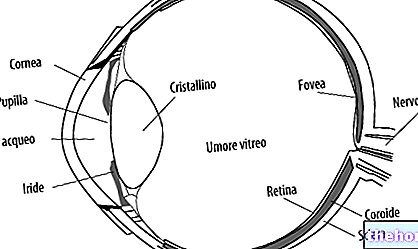
Retinitis pigmentosa je retinálna dystrofia charakterizovaná postupnou stratou fotoreceptorov a dysfunkciou pigmentového epitelu. To znamená, že sietnica postupne znižuje svoju schopnosť prenášať zrakové informácie do mozgu optickým nervom.
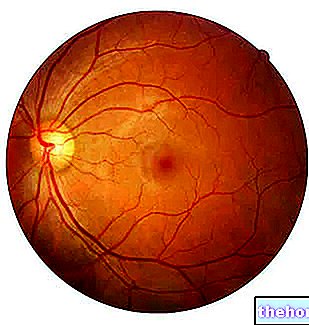
Patologický proces začína zmenami sietnicového pigmentového epitelu. Ako postupuje retinitis pigmentosa, dochádza k rednutiu ciev, ktoré zásobujú sietnicu a podliehajú atrofii. Po vyšetrení fundusu sú vizuálne zistiteľné charakteristické ložiská. Sietnicový pigment ( odtiaľ názov choroby). Atrofické zmeny a poškodenia môžu zahŕňať aj zrakový nerv a postupne odumierajú fotosenzitívne bunky sietnice.
Pacienti postihnutí retinitis pigmentosa majú spočiatku problémy so zrakom, najmä v zle osvetlenom prostredí, a sťažujú sa na zúženie periférneho zorného poľa. Centrálne videnie je ušetrené až do neskorších štádií ochorenia a konečný výsledok sa môže dramaticky líšiť: mnoho ľudí s retinitis pigmentosa si po celý život zachováva obmedzené videnie, zatiaľ čo iní úplne strácajú zrak.
Retinitis pigmentosa je dedičné ochorenie, ktoré je spôsobené predovšetkým genetickými zmenami prenášanými od jedného alebo oboch rodičov. Typ genetického defektu určuje, ktoré sietnicové bunky sa na poruche najviac podieľajú, a umožňuje z klinického hľadiska rozlíšiť rôzne stavy. Doteraz bolo identifikovaných viac ako 50 rôznych genetických defektov implikovaných pri retinitis pigmentosa. Abnormality môžu byť prenášané z rodičov na potomstvo prostredníctvom jedného z troch dedičných vzorcov: autozomálne recesívny, autozomálne dominantný alebo heterozomálne recesívny (spojený s X alebo X).
Príznaky
Ďalšie informácie: Príznaky sietnicovej pigmentácie
Retinitis pigmentosa sa zvyčajne vyskytuje u dospievajúcich a mladých dospelých. Príznaky sa často objavujú vo veku 10 až 30 rokov, ale diagnózu je možné stanoviť v ranom detstve alebo oveľa neskôr v živote.
Počiatočné symptómy retinitis pigmentosa môžu zahŕňať:
- Obtiažne videnie v noci (nočná slepota) alebo pri slabom osvetlení
- Pomalé prispôsobovanie sa z videnia v tme na svetlo, a naopak;
- Zúženie zorného poľa a strata periférneho videnia;
- Citlivosť na svetlo a oslnenie.
Niektoré príznaky závisia od typu zapojených fotoreceptorov. Tyče sú zodpovedné za čierne a biele videnie, zatiaľ čo kužele vám umožňujú rozlíšiť farby.
Vo väčšine prípadov retinitis pigmentosa sú najskôr zapojené tyčinky. V rýchlo sa vyvíjajúcich formách však môžu byť kužele postihnuté aj v počiatočnom štádiu.
Tyčinky sú koncentrované vo vonkajších častiach sietnice a sú aktivované slabým svetlom, takže ich degenerácia ovplyvňuje periférne a nočné videnie. Ak sú zahrnuté kužele, je možné zažiť stratu vnímania farieb a centrálneho videnia.
Prevaha zapojených fotoreceptorov je daná konkrétnym defektom prítomným v genetickej výbave pacienta.
Prvým symptómom retinitis pigmentosa je často nočná slepota (alebo noktalopia). Niektorí ľudia zisťujú, že potrebujú stále viac času na prispôsobenie sa rozdielom vo svetle, keď sa presúvajú z dobre osvetlenej oblasti do tmavšej. Typická forma straty zraku spôsobuje zúženie periférneho videnia (tunelové alebo teleskopické videnie); tento vzor sa nazýva kruhový skotóm. Niekedy tento jav môže chýbať v počiatočných štádiách, ale je zaznamenaný, keď jednotlivec často zakopáva o predmety alebo je účastníkom dopravnej nehody. Keď strata zraku zahŕňa centrálnu oblasť sietnice (nazývanú aj makulárna dystrofia), pacienti majú problémy s čítaním a podrobnou prácou, ktorá si vyžaduje koncentráciu na jeden predmet, ako je prevlečenie vlákna niťou cez ucho ihly Mnoho pacientov uvádza, že videli záblesky svetla (fotopsia), často popisované ako malé, blikajúce a trblietavé svetlá.
Rýchlosť progresie ochorenia a stupeň straty zraku sa líšia od človeka k človeku. Niektoré extrémne prípady sa môžu rýchlo vyvinúť do dvoch desaťročí, iné pomalým kurzom, ktorý nikdy nevedie k úplnej slepote. Skorý nástup sa vyskytuje u závažnejších foriem retinitis pigmentosa, zatiaľ čo u pacientov s miernejšími stavmi (napr. Autozomálne dominantnými) sa choroba môže rozvinúť v piatej alebo šiestej dekáde života. V rodinách s X-retinitis pigmentosa sú muži postihnutí častejšie než ženy a vážnejšie; ženy na druhej strane prenášajú genetické vlastnosti (nesú zmenený gén na chromozóme X) a symptómy poruchy prejavujú menej často.
Komplikácie
Retinitis pigmentosa bude pokračovať, aj keď pomaly. Úplná slepota je však zriedkavá, ale môže dôjsť k významnému zníženiu periférneho a centrálneho videnia.
U pacientov s retinitis pigmentosa sa v útlom veku často vyvinie opuch sietnice (makulárny edém) alebo katarakta. Tieto komplikácie je možné liečiť, ak zasahujú do videnia.
Súvisiace choroby
Bežne nemá pacient s retinitis pigmentosa žiadne iné poruchy a v tomto prípade hovoríme o „nesyndrómovej“ alebo jednoduchej retinitis pigmentosa. Niekoľko syndrómov má však s týmto očným ochorením určité klinické príznaky; najčastejším je Usherov syndróm, ktorý postihuje približne 10-30% všetkých pacientov s retinitis pigmentosa a je spojený so súčasnou vrodenou alebo progresívnou stratou sluchu. Pri Leberovej vrodenej amauróze môžu deti počas prvých šiestich mesiacov života oslepnúť alebo takmer oslepnúť. Medzi ďalšie choroby súvisiace s retinitis pigmentosa patrí Bardet-Biedlov syndróm a Refsumova choroba.
Príčiny
Ochorenie môže byť spôsobené mnohými genetickými poruchami: v skutočnosti existuje niekoľko génov, ktoré v prípade zmeny môžu spôsobiť fenotyp retinitis pigmentosa. Tieto bežne kódujú proteíny zahrnuté v transdukčnej kaskáde, ktorá umožňuje videnie, faktory transkripciu buniek. (ktoré odosielajú chybné správy do sietnicových buniek) alebo pre prvky, ktoré tvoria štruktúru fotoreceptorov. Dedičné génové mutácie sú v bunkách prítomné od okamihu počatia; medzi bežné abnormality patria mutácie génov RP1 (pri retinitis pigmentosa-1, autozomálne dominantné) , RHO (RP4, autozomálne dominantná) a RDS (RP7, autozomálne dominantná). Dedičné príčiny retinitis pigmentosa sú zriedkavé, ale možnosť nájsť izolovaný prípad (spontánna mutácia), v ktorom nie je prítomná rodinná anamnéza choroba.