Účinné látky: sunitinib
SUTENT 12,5 mg tvrdé kapsuly
SUTENT 25 mg tvrdé kapsuly
SUTENT 37,5 mg tvrdé kapsuly
SUTENT 50 mg tvrdé kapsuly
Indikácie Prečo sa používa Sutent? Načo to je?
Sutent obsahuje účinnú látku sunitinib, ktorá je inhibítorom proteínkinázy. Používa sa na liečbu rakoviny prevenciou aktivity špecifickej skupiny proteínov, o ktorých je známe, že sa podieľajú na raste a šírení rakovinotvorných buniek.
Sutent vám predpíše iba lekár, ktorý má skúsenosti s používaním liekov proti rakovine.
Sutent sa používa na liečbu dospelých s nasledujúcimi druhmi rakoviny:
- Gastrointestinálna stromálna rakovina (GIST), typ rakoviny žalúdka a čriev, v prípadoch, keď imatinib (iný protirakovinový liek) už nefunguje alebo ho už nemožno užívať.
- Metastatická rakovina obličiek (MRCC), typ rakoviny obličiek, ktorá sa rozšírila do iných častí tela.
- Pankreatické neuroendokrinné nádory (pNET) (nádory buniek pankreasu produkujúcich hormóny), ktoré progredujú alebo sú neresekovateľné
. Ak si nie ste istí, ako účinkuje Sutent alebo prečo vám bol tento liek predpísaný, opýtajte sa svojho lekára.
Kontraindikácie Keď sa Sutent nemá používať
Neužívajte Sutent:
- Ak ste alergický na sunitinib alebo na ktorúkoľvek z ďalších zložiek tohto lieku (uvedených v časti 6).
Opatrenia pri používaní Čo potrebujete vedieť predtým, ako užijete Sutent
Predtým, ako začnete užívať Sutent, povedzte to svojmu lekárovi:
- Ak máte vysoký krvný tlak. Sutent môže spôsobiť zvýšenie vášho krvného tlaku. Váš lekár vám môže počas užívania Sutentu kontrolovať krvný tlak a v prípade potreby bude musieť užívať lieky na zníženie krvného tlaku.
- Ak máte alebo ste mali poruchy krvi, problémy s krvácaním alebo modriny. Liečba Sutentom môže predstavovať zvýšené riziko krvácania, zmien v počte určitých krviniek, ktorých nedostatok vedie k anémii alebo ovplyvňuje zrážanlivosť krvi. Riziko krvácania môže byť vyššie, ak užívate warfarín alebo acenokumarol, lieky na riedenie krvi na prevenciu krvných zrazenín. Povedzte svojmu lekárovi, ak počas užívania Sutentu zaznamenáte akékoľvek krvácanie.
- Ak máte problémy so srdcom. Sutent môže spôsobiť srdcové problémy. Ak sa cítite veľmi unavení, dýchavičíte alebo máte opuchnuté nohy a členky, povedzte to svojmu lekárovi.
- Ak zaznamenáte abnormálne zmeny srdcového rytmu. Sutent môže spôsobiť zmeny srdcového rytmu. Počas liečby Sutentom vám môže lekár urobiť elektrokardiogram, aby zhodnotil rozsah týchto zmien. Ak sa počas užívania Sutentu cítite závraty, mdloby alebo máte abnormálne srdcové tepny, povedzte to svojmu lekárovi.
- Ak ste nedávno mali problémy s krvnými zrazeninami v žilách a / alebo tepnách (typy krvných ciev), vrátane cievnej mozgovej príhody, srdcového infarktu, embólie alebo trombózy. Ak sa u vás objavia príznaky ako zvieranie alebo bolesť na hrudníku, bolesť paží, chrbta, krku alebo čeľuste, dýchavičnosť, znecitlivenie alebo slabosť na jednej strane tela, chvenie pri chôdzi, bolesť počas liečby Sutentom, ihneď kontaktujte svojho lekára. alebo závraty.
- Ak máte problémy so štítnou žľazou. Sutent môže spôsobiť problémy so štítnou žľazou. Povedzte svojmu lekárovi, ak sa počas užívania Sutentu ľahšie unavíte, zvyčajne sa cítite chladnejšie ako ostatní ľudia alebo sa vám zníži hlas. Pred užívaním Sutentu a pravidelne počas užívania lieku by ste mali skontrolovať funkciu štítnej žľazy. Ak štítna žľaza neprodukuje dostatok hormónu štítnej žľazy, môže byť potrebné vziať si náhradný hormón štítnej žľazy.
- Ak máte alebo ste mali problémy s pankreasom alebo žlčníkom. Povedzte svojmu lekárovi, ak sa u vás prejaví niektorý z nasledujúcich prejavov a symptómov: bolesť žalúdka (hornej časti brucha), nevoľnosť, vracanie a horúčka.To môže byť spôsobené zápalom pankreasu alebo žlčníka.
- Ak máte alebo ste niekedy mali problémy s pečeňou. Povedzte svojmu lekárovi, ak sa u vás počas liečby Sutentom prejaví niektorý z nasledujúcich prejavov a príznakov problémov s pečeňou: svrbenie, zožltnutie pokožky alebo očí, tmavý moč a bolesť alebo nepríjemné pocity v pravej hornej časti žalúdka. Váš lekár by mal vykonať testy. na kontrolu funkcie pečene pred a počas liečby Sutentom a podľa klinického stavu.
- Ak máte alebo ste mali problémy s obličkami. Lekár bude sledovať funkciu obličiek.
- Ak sa chystáte na operáciu alebo ste nedávno podstúpili operáciu. Sutent môže ovplyvniť spôsob hojenia rán. Ak sa chystáte na operáciu, spravidla budete musieť prestať používať Sutent. Váš lekár rozhodne, kedy znova začať liečbu Sutentom.
- Pred začatím liečby Sutentom sa odporúča vykonať zubnú prehliadku.
- ak máte alebo ste mali bolesť v ústach, zuboch a / alebo čeľusti, opuch alebo vredy v ústach, necitlivosť alebo pocit ťažkosti v čeľusti alebo uvoľnenie zubov, ihneď to povedzte svojmu lekárovi a zubárovi.
- ak podstupujete invazívne zubné ošetrenie alebo zubný chirurgický zákrok, povedzte svojmu lekárovi, že sa liečite Sutentom, najmä ak súčasne užívate alebo ste v minulosti užívali intravenózne bisfosfonáty.Bisfosfonáty sú lieky používané na prevenciu kostných komplikácií, ktoré mohli byť predpísané pre iný zdravotný problém.
- Ak máte alebo ste niekedy mali poruchy kože a podkožného tkaniva. Počas liečby týmto liekom sa môže objaviť „gangrenózna pyodermia“ (bolestivá ulcerácia kože) alebo „nekrotizujúca fasciitída“ (rýchlo sa šíriaca „infekcia kože / mäkkých tkanív“. Prerušenie liečby. Závažné kožné reakcie (Stevens-Johnson syndróm, toxická epidermálna nekrolýza, multiformný erytém) boli hlásené pri použití sunitinibu, spočiatku sa objavujúceho na trupu ako načervenalé terčovité škvrny alebo kruhové škvrny, často s pľuzgiermi v strede. Reakcia môže viesť k rozsiahlym pľuzgierom alebo odlupovaniu kože a môže byť smrteľná. Ak sa u vás objaví vyrážka alebo niektorý z týchto kožných symptómov, ihneď sa poraďte s lekárom.
- Ak máte alebo ste mali záchvaty. Čo najskôr povedzte svojmu lekárovi, ak máte vysoký krvný tlak, bolesť hlavy, stratu zraku.
- Ak máte cukrovku. U diabetických pacientov by sa mala pravidelne kontrolovať hladina cukru v krvi, aby sa zistilo, či je potrebné zmeniť dávkovanie liekov na cukrovku, aby sa minimalizovalo riziko nízkej hladiny cukru v krvi.
Deti a dospievajúci
Sutent nie je indikovaný u pacientov mladších ako 18 rokov. Sutent sa neskúmal u detí a dospievajúcich.
Interakcie Ktoré lieky alebo potraviny môžu meniť účinok Sutentu
Ak teraz užívate alebo ste v poslednom čase užívali, či práve budete užívať ďalšie lieky, povedzte to svojmu lekárovi alebo lekárnikovi.
Niektoré lieky môžu meniť hladiny Sutentu v tele. Ak užívate lieky, ktoré obsahujú nasledujúce účinné látky, oznámte to svojmu lekárovi:
- ketokonazol, itrakonazol - používané na liečbu plesňových infekcií
- erytromycín, klaritromycín, rifampicín - používané na liečbu infekcií
- ritonavir - používa sa na liečbu AIDS
- dexametazón - kortikosteroid používaný na niekoľko stavov
- fenytoín, karbamazepín, fenobarbital - používajú sa na liečbu epilepsie a iných neurologických stavov
- bylinné prípravky obsahujúce ľubovník bodkovaný (Hypericum perforatum) - používajú sa na liečbu depresie a úzkosti
Sutent s jedlom a nápojmi
Počas liečby Sutentom sa treba vyhýbať príjmu grapefruitovej šťavy.
Upozornenia Je dôležité vedieť, že:
Tehotenstvo a dojčenie
Ak ste tehotná alebo máte podozrenie, že ste tehotná, povedzte to svojmu lekárovi.
Sutent sa nemá používať počas tehotenstva, pokiaľ to nie je úplne nevyhnutné. Váš lekár sa s vami porozpráva o možných rizikách liečby Sutentom počas tehotenstva.
Ak je tehotenstvo možné, musíte počas liečby Sutentom používať spoľahlivú metódu antikoncepcie.
Ak dojčíte, povedzte to svojmu lekárovi. Počas liečby Sutentom by ste nemali dojčiť.
Vedenie vozidla a obsluha strojov
Ak pociťujete závrat alebo neobvyklú únavu, buďte obzvlášť opatrní pri vedení vozidla alebo obsluhe strojov.
Dávka, spôsob a čas podávania Ako používať Sutent: Dávkovanie
Vždy užívajte tento liek presne tak, ako vám povedal váš lekár.
Ak máte pochybnosti, poraďte sa so svojim lekárom. Váš lekár vám predpíše správnu dávku podľa typu rakoviny, ktorú musíte liečiť. Ak sa liečite na GIST alebo MRCC, zvyčajná dávka je 50 mg jedenkrát denne, ktorá sa má užívať 28 dní (4 týždne), po ktorých nasleduje 14 dní (2 týždne) odpočinku (bez liekov), v 6 -týždňových cykloch. Ak sa liečite na pNET, zvyčajná dávka je 37,5 mg jedenkrát denne bez prestávky. Váš lekár určí dávku, ktorú potrebujete, a kedy ukončiť liečbu Sutentom. Sutent sa môže užívať s jedlom alebo bez jedla.
Predávkovanie Čo robiť, ak ste užili príliš veľa Sutentu
Ak užijete viac Sutentu, ako máte
Ak ste omylom užili príliš veľa kapsúl, ihneď to povedzte svojmu lekárovi. Môže byť potrebná lekárska pomoc
Ak zabudnete užiť Sutent
Neužívajte dvojnásobnú dávku, aby ste nahradili vynechanú dávku.
Vedľajšie účinky Aké sú vedľajšie účinky lieku Sutent
Tak ako všetky lieky, aj tento liek môže spôsobovať vedľajšie účinky, hoci sa neprejavia u každého.
Ak sa u vás vyskytne ktorýkoľvek z týchto závažných vedľajších účinkov, ihneď kontaktujte svojho lekára (pozri tiež Čo potrebujete vedieť predtým, ako užijete Sutent):
Problémy so srdcom. Ak sa cítite veľmi unavení, dýchavičíte alebo máte opuchnuté nohy a členky, povedzte to svojmu lekárovi. Môžu to byť príznaky srdcových problémov, ako je srdcové zlyhanie a problémy so srdcovým svalom (kardiomyopatia).
Problémy s pľúcami alebo dýchaním. Povedzte svojmu lekárovi, ak pocítite kašeľ, bolesť na hrudníku, náhly nástup dýchavičnosti alebo vykašliavanie krvi. Môžu to byť príznaky pľúcnej embólie, ku ktorej dochádza vtedy, keď krvné zrazeniny putujú do pľúc.
Problémy s obličkami. Ak sa u vás vyskytne „zmenená frekvencia alebo absencia močenia, čo môžu byť príznaky“ zlyhania obličiek, povedzte to svojmu lekárovi.
Krvácajúca. Ak sa u vás počas užívania Sutentu vyskytne niektorý z nasledujúcich príznakov alebo závažný problém s krvácaním, ihneď to povedzte svojmu lekárovi: opuchnutý, bolestivý žalúdok (brucho); vracanie s krvou; tmavé, lepkavé stolice; bolesť hlavy alebo zmeny duševného stavu, vykašliavanie krvi alebo sputa krvou z pľúc alebo dýchacích ciest.
Zničenie nádoru spôsobujúce perforáciu čreva. Povedzte svojmu lekárovi, ak máte silné bolesti čriev, horúčku, nevoľnosť, vracanie, krv v stolici alebo zmeny vo vyprázdňovaní.
Ďalšie vedľajšie účinky, ktoré sa môžu vyskytnúť pri Sutente, sú:
Veľmi časté vedľajšie účinky (môžu postihnúť viac ako 1 z 10 ľudí)
- Zníženie počtu krvných doštičiek, červených krviniek a / alebo bielych krviniek (napr. Neutrofilov).
- Lapanie po dychu
- Vysoký krvný tlak.
- Nadmerná únava, strata sily.
- Opuch spôsobený tekutinou pod kožou a okolo očí, hlboká alergická vyrážka.
- Bolesť / podráždenie úst, bolestivosť / zápal / sucho v ústach, poruchy chuti, podráždenie žalúdka, nevoľnosť, vracanie, hnačka, zápcha, bolesť / opuch brucha, strata / zníženie chuti do jedla.
- Znížená činnosť štítnej žľazy (hypotyreóza).
- Závraty
- Bolesť hlavy
- Krvácanie z nosa.
- Bolesti chrbta, kĺbov.
- Bolesť v rukách a nohách.
- Zožltnutie pokožky / zmena farby pokožky, nadmerná pigmentácia pokožky, zmena farby vlasov, vyrážka na dlaniach a chodidlách, vyrážka, suchá koža.
- Kašeľ
- Horúčka.
- Ťažké zaspávanie.
Časté vedľajšie účinky (môžu postihnúť 1 až 10 zo 100 ľudí)
- Tvorba zrazeniny v cievach.
- Nedostatočné prekrvenie srdcového svalu v dôsledku upchatia alebo zúženia koronárnych artérií.
- Bolesť v hrudi.
- Znížené množstvo krvi čerpanej srdcom.
- Zadržiavanie tekutín aj v okolí pľúc.
- Infekcie.
- Znížená hladina cukru v krvi. Ak sa u vás prejavia príznaky a príznaky nízkej hladiny cukru v krvi: Čo najskôr informujte svojho lekára, ak pocítite únavu, búšenie srdca, potenie, hlad a stratu vedomia.
- Strata bielkovín v moči, ktorá niekedy vedie k opuchu.
- Chrípkový syndróm.
- Abnormálne krvné testy vrátane hladín pečeňových a pankreatických enzýmov.
- Vysoká hladina kyseliny močovej v krvi.
- Hemoroidy, bolesť konečníka, krvácanie z ďasien, problémy s prehĺtaním alebo neschopnosť prehĺtať.
- Pálivý alebo bolestivý pocit v jazyku, zápal sliznice tráviaceho traktu, prebytočný plyn v žalúdku alebo črevách.
- Strata váhy.
- Muskuloskeletálna bolesť (bolesť svalov a kostí), svalová slabosť, svalová únava, bolesť svalov, svalové kŕče.
- Suchosť nosa, upchatý nos.
- Nadmerné trhanie.
- Zmeny citlivosti pokožky, suchá koža, svrbenie, odlupovanie a zápaly pokožky, pľuzgiere, akné, zmena farby nechtov, vypadávanie vlasov.
- Abnormálne pocity v končatinách.
- Nadmerné zníženie / zvýšenie citlivosti, najmä na dotyk.
- Pálenie v žalúdku.
- Dehydratácia.
- Sčervenanie tváre.
- Zmena farby moču.
- Depresia.
- Zimnica.
Menej časté vedľajšie účinky (môžu postihnúť 1 až 10 z 1 000 ľudí)
- Infekcie mäkkých tkanív, vrátane anogenitálnej oblasti, potenciálne život ohrozujúce.Ak pocítite príznaky infekcie okolo kožnej rany, vrátane horúčky, bolesti, začervenania, opuchu alebo odtoku hnisu alebo krvi, ihneď kontaktujte svojho lekára.
- Mŕtvica.
- Infarkt spôsobený prerušeným alebo zníženým prívodom krvi do srdca.
- Zmeny v elektrickej aktivite srdca alebo zmenený srdcový rytmus.
- Tekutina okolo srdca (perikardiálny výpotok).
- Pečeňová insuficiencia.
- Bolesť žalúdka (brucha) spôsobená „zápalom pankreasu.
- Zničenie nádoru spôsobujúce perforáciu čreva.
- Zápal (opuch a začervenanie) žlčníka s pridruženými kameňmi alebo bez nich.
- Abnormálny komunikačný kanál medzi dvoma telesnými dutinami alebo s pokožkou.
- Bolesť v ústach, zuboch a / alebo čeľusti, opuch alebo podráždenie v ústach, necitlivosť alebo pocit ťažkosti v čeľusti alebo uvoľnenie zubov. Môžu to byť príznaky a symptómy poranenia čeľustnej kosti (osteonekróza). Ak sa u vás prejaví ktorýkoľvek z týchto prejavov a symptómov, ihneď to oznámte svojmu lekárovi a zubnému lekárovi.
- Nadmerná produkcia hormónov štítnej žľazy má za následok zvýšený metabolizmus. Problémy s hojením rán po operácii.
- Zvýšenie svalového enzýmu v krvi (kreatínfosfokináza).
- Nevhodná a nadmerná reakcia na alergény.
Zriedkavé vedľajšie účinky (môžu postihnúť 1 až 10 z 10 000 ľudí)
- Závažné reakcie kože a / alebo slizníc (Stevensov-Johnsonov syndróm, toxická epidermálna nekrolýza, multiformný erytém).
- Syndróm nádorovej lýzy (TLS) - TLS zahŕňa súbor metabolických komplikácií, ktoré sa môžu vyskytnúť počas liečby rakoviny. Sú spôsobené produktmi rozkladu postihnutých rakovinových buniek a môžu zahŕňať: nevoľnosť, dýchavičnosť, nepravidelný srdcový tep, svalové kŕče, kŕče, zakalený moč a únavu spojenú s abnormálnymi výsledkami laboratórnych testov (vysoké hladiny draslíka, kyseliny močovej a kyseliny fosforečnej a nízke hladiny vápnika v krvi), ktoré môžu viesť k zmenám funkcie obličiek a akútnemu zlyhaniu obličiek.
- Abnormálne rozpadnutie svalov, ktoré môže spôsobiť problémy s obličkami (rabdomyolýza).
- Zhoršená funkcia mozgu, ktorá môže viesť k rôznym symptómom, ako je bolesť hlavy, zmätenosť, záchvaty a strata zraku (syndróm zadnej reverzibilnej leukoencefalopatie).
- Bolestivé vredy na koži (gangrenózna pyodermia).
- Zápal pečene (hepatitída).
- Zápal štítnej žľazy.
Hlásenie vedľajších účinkov
Ak sa u vás vyskytne akýkoľvek vedľajší účinok, obráťte sa na svojho lekára. To sa týka aj akýchkoľvek vedľajších účinkov, ktoré nie sú uvedené v tejto písomnej informácii. Vedľajšie účinky môžete hlásiť aj priamo prostredníctvom národného systému hlásenia uvedeného v Prílohe V. Hlásením vedľajších účinkov môžete prispieť k získaniu ďalších informácií o bezpečnosti tohto lieku.
Expirácia a retencia
- Tento liek uchovávajte mimo dohľadu a dosahu detí.
- Nepoužívajte tento liek po dátume exspirácie, ktorý je uvedený na škatuli a na štítku po „EXP“. Dátum exspirácie sa vzťahuje na posledný deň v mesiaci.
- Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
- Nepoužívajte tento liek, ak spozorujete, že balenie je poškodené alebo javí známky nedovoleného zaobchádzania.
Nelikvidujte lieky odpadovou vodou alebo domovým odpadom. Nepoužitý liek vráťte do lekárne. Pomáha to chrániť životné prostredie.
Čo Sutent obsahuje
Sutent 12,5 mg tvrdé kapsuly
Účinnou zložkou lieku je sunitinib. Každá kapsula obsahuje sunitinib malát zodpovedajúci 12,5 mg sunitinibu.
Ďalšie zložky sú:
- Obsah kapsuly: manitol (E421), sodná soľ kroskarmelózy, povidón (K-25) a magnéziumstearát.
- Obal kapsuly: želatína, červený oxid železitý (E172) a oxid titaničitý (E171).
- Atrament: šelak, propylénglykol, hydroxid sodný, povidón a oxid titaničitý (E171).
Sutent 25 mg tvrdé kapsuly
Účinnou látkou je sunitinib. Každá kapsula obsahuje sunitinib malát zodpovedajúci 25 mg sunitinibu.
Ďalšie zložky sú:
- Obsah kapsuly: manitol (E421), sodná soľ kroskarmelózy, povidón (K-25) a magnéziumstearát.
- Obal kapsuly: želatína, oxid titaničitý (E171), žltý oxid železitý (E172), červený oxid železitý (E172), čierny oxid železitý (E172).
- Atrament: šelak, propylénglykol, hydroxid sodný, povidón a oxid titaničitý (E171)
Sutent 37,5 mg tvrdé kapsuly
Účinnou látkou je sunitinib. Každá kapsula obsahuje sunitinib malát zodpovedajúci 37,5 mg sunitinibu.
Ďalšie zložky sú:
- Obsah kapsuly: manitol (E421), sodná soľ kroskarmelózy, povidón (K-25) a magnéziumstearát.
- Obal kapsuly: želatína, oxid titaničitý (E171), žltý oxid železitý (E172).
- Atrament: šelak, propylénglykol, hydroxid draselný, čierny oxid železitý (E172).
Sutent 50 mg tvrdé kapsuly
Účinnou zložkou lieku je sunitinib. Každá kapsula obsahuje sunitinib malát zodpovedajúci 50 mg sunitinibu.
Ďalšie zložky sú:
- Obsah kapsuly: manitol (E421), sodná soľ kroskarmelózy, povidón (K-25) a magnéziumstearát.
- Obal kapsuly: želatína, oxid titaničitý (E171), žltý oxid železitý (E172), červený oxid železitý (E172) a čierny oxid železitý (E172).
- Atrament: šelak, propylénglykol, hydroxid sodný, povidón a oxid titaničitý (E171).
Opis toho, ako Sutent vyzerá a obsah balenia
Sutent 12,5 mg je dostupný vo forme tvrdých želatínových kapsúl s oranžovým vrchnákom a telom, s bielym atramentom „Pfizer“ na viečku a „STN 12,5 mg“ na tele, ktoré obsahujú farebné granule. Žltooranžové.
Sutent 25 mg je dostupný vo forme tvrdých želatínových kapsúl s karamelovým viečkom a oranžovým telom, s bielym atramentom „Pfizer“ na vrchnáku a „STN 25 mg“ na tele, ktoré obsahujú farebné granule. Žltooranžové.
Sutent 37,5 mg je dostupný vo forme tvrdých želatínových kapsúl so žltým vrchnákom a telom, s čiernym atramentom „Pfizer“ na viečku a „STN 37,5 mg“ na tele, ktoré obsahujú farebné granule. Žltooranžové.
Sutent 50 mg je dostupný vo forme tvrdých želatínových kapsúl s viečkom a telom karamelovej farby, s viečkom „Pfizer“ bielym atramentom a s telom „STN 50 mg“ na tele, ktoré obsahujú žltooranžové granule. Je dostupný vo fľašiach s 30 kapsulami a perforovaných blistroch s jednotlivou dávkou obsahujúcich 28 x 1 kapsulu.
Na trh nemusia byť uvedené všetky veľkosti balenia.
Zdrojový leták: AIFA (Talianska agentúra pre lieky). Obsah zverejnený v januári 2016. Súčasné informácie nemusia byť aktuálne.
Aby ste mali prístup k najaktuálnejšej verzii, odporúča sa navštíviť webovú stránku AIFA (Talianska agentúra pre lieky). Vylúčenie zodpovednosti a užitočné informácie.
01.0 NÁZOV LIEKU
SUTENT 12,5 MG TVRDÉ Kapsle
02.0 KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá kapsula obsahuje sunitinib malát, čo zodpovedá 12,5 mg sunitinibu.
Úplný zoznam pomocných látok, pozri časť 6.1.
03.0 LIEKOVÁ FORMA
Tvrdá kapsula.
Želatínové kapsuly s oranžovým vrchnákom a telom, označené bielym atramentom „Pfizer“ na viečku, „STN 12,5 mg“ na tele a obsahujúce žltooranžové granule.
04.0 KLINICKÉ INFORMÁCIE
04.1 Terapeutické indikácie
Stromálny nádor gastrointestinálneho traktu (GIST)
SUTENT je indikovaný na liečbu neresekovateľného a / alebo metastatického gastrointestinálneho stromálneho karcinómu (GIST) u dospelých po zlyhaní liečby imatinibom v dôsledku rezistencie alebo intolerancie.
Metastatický karcinóm obličkových buniek (MRCC)
SUTENT je indikovaný na liečbu pokročilého / metastatického karcinómu obličkových buniek (MRCC) u dospelých.
Pankreatické neuroendokrinné nádory (pNET)
SUTENT je indikovaný na liečbu dobre diferencovaných, neresekovateľných alebo metastatických pankreatických neuroendokrinných nádorov (pNET) pri progresii ochorenia u dospelých.
Skúsenosti so SUTENTOM ako liekom prvej voľby sú obmedzené (pozri časť 5.1).
04.2 Dávkovanie a spôsob podávania
Terapiu sunitinibom má začať lekár so skúsenosťami s podávaním protirakovinových liekov.
Dávkovanie
Pre GIST a MRCC je odporúčaná dávka SUTENTu 50 mg, ktorá sa má užívať perorálne jedenkrát denne počas 4 po sebe nasledujúcich týždňov, po ktorých nasledujú 2 týždne prestávky (schéma 4/2), aby sa celý cyklus absolvoval 6 týždňov.
Pre pNET je odporúčaná dávka SUTENTu 37,5 mg, ktorá sa má užívať perorálne jedenkrát denne bez plánovanej prestávky.
Úprava dávky
Bezpečnosť a znášanlivosť
V prípade GIST a MRCC je možné dávku upravovať v prírastkoch 12,5 mg na základe individuálnej bezpečnosti a znášanlivosti pacienta. Denná dávka by nemala prekročiť 75 mg ani by nemala byť znížená pod 25 mg.
V prípade pNET je možné dávkovanie upravovať v prírastkoch 12,5 mg na základe individuálnej bezpečnosti a znášanlivosti pacienta. Maximálna dávka podaná v štúdii pNET fázy 3 bola 50 mg denne.
Na základe bezpečnosti a znášanlivosti jednotlivého pacienta môže byť potrebné prerušiť príjem niektorých dávok.
Inhibítory / induktory CYP3A4
Je potrebné vyhnúť sa súbežnému podávaniu sunitinibu so silnými induktormi CYP3A4, ako je rifampicín (pozri časti 4.4 a 4.5). Ak to nie je možné, dávku sunitinibu možno bude potrebné zvýšiť v prírastkoch 12,5 mg (až do 87,5 mg / deň pre GIST a MRCC alebo 62,5 mg / deň pre pNET) na základe starostlivého monitorovania znášanlivosti.
Je potrebné vyhnúť sa súbežnému podávaniu sunitinibu so silnými inhibítormi CYP3A4, ako je ketokonazol (pozri časti 4.4 a 4.5). Ak to nie je možné, dávku sunitinibu možno bude potrebné znížiť na minimálnu dávku 37,5 mg denne pre GIST a MRCC alebo 25 mg denne pre pNET na základe starostlivého monitorovania znášanlivosti.
Má sa zvážiť výber alternatívneho súbežne podávaného lieku, ktorý nemá žiadny alebo má minimálny potenciál indukovať alebo inhibovať CYP3A4.
Špeciálne populácie
Pediatrická populácia
Bezpečnosť a účinnosť sunitinibu u pacientov mladších ako 18 rokov nebola stanovená.
Nie sú k dispozícii žiadne údaje.
Neexistuje žiadna indikácia pre špecifické použitie sunitinibu u detí od narodenia do 6 rokov na liečbu neresekovateľného a / alebo metastatického GIST po zlyhaní liečby imatinibom v dôsledku rezistencie alebo intolerancie. Neexistuje žiadna indikácia pre špecifické použitie sunitinibu v pediatrickej populácii pri liečbe MRCC a pri liečbe dobre diferencovaných, neresekovateľných alebo metastatických pNET v progresii ochorenia.
Použitie sunitinibu v pediatrickej populácii sa neodporúča.
Starší pacienti (≥ 65 rokov)
Približne jedna tretina pacientov zaradených do klinických skúšaní, ktorí dostávali sunitinib, boli vo veku 65 rokov alebo starší.Nezaznamenali sa žiadne významné rozdiely v bezpečnosti a účinnosti medzi mladšími a staršími subjektmi.
Poškodenie funkcie pečene
Pri podávaní sunitinibu pacientom s miernym alebo stredne ťažkým poškodením funkcie pečene (Childovo-Pughovo štádium A a B) nie je potrebná žiadna úprava úvodnej dávky. Použitie sunitinibu u osôb s ťažkou poruchou funkcie pečene (Childovo-Pughovo štádium C) sa neskúmalo, preto sa jeho použitie u pacientov s poruchou funkcie pečene neodporúča (pozri časť 5.2).
Porucha funkcie obličiek
Pri podávaní sunitinibu pacientom s poruchou funkcie obličiek (stredne ťažká až ťažká) alebo s ochorením obličiek v konečnom štádiu (ESRD) podstupujúcich hemodialýzu nie je potrebná žiadna úprava úvodnej dávky. Následné úpravy dávky sa majú vykonať na základe bezpečnosti a znášanlivosti jednotlivého pacienta (pozri časť 5.2).
Spôsob podávania
SUTENT je na perorálne podanie. Môže sa užívať s jedlom alebo bez jedla.
Ak sa dávka neužije, ďalšia dávka sa nemá podať. Nasledujúci deň má pacient užiť zvyčajnú predpísanú dávku.
04.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
04.4 Špeciálne upozornenia a vhodné opatrenia pri používaní
Je potrebné vyhnúť sa súbežnému podávaniu so silnými induktormi CYP3A4, pretože môže znížiť plazmatickú koncentráciu sunitinibu (pozri časti 4.2 a 4.5).
Je potrebné vyhnúť sa súbežnému podávaniu so silnými inhibítormi CYP3A4, pretože môže zvýšiť plazmatickú koncentráciu sunitinibu (pozri časti 4.2 a 4.5).
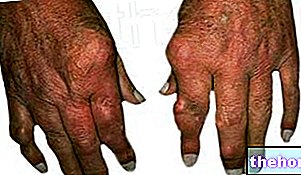
Poruchy kože a tkaniva
Odfarbenie kože, ktoré môže byť dôsledkom farby účinnej látky (žltá), je veľmi častou nežiaducou reakciou, ktorá sa vyskytuje približne u 30% pacientov. Pacientov treba upozorniť, že počas liečby sunitinibom môžu zafarbiť vlasy alebo môže sa objaviť aj koža. Ďalšie možné dermatologické účinky môžu zahŕňať suchosť, zhrubnutie alebo popraskanie pokožky, pľuzgiere alebo občasnú vyrážku na dlaniach alebo chodidlách.
Vyššie uvedené reakcie neboli kumulatívne, boli spravidla reverzibilné a zvyčajne neviedli k prerušeniu liečby. Boli hlásené prípady gangrenóznej pyodermie, ktoré sú po prerušení liečby spravidla reverzibilné. Boli hlásené závažné kožné reakcie, vrátane prípadov multiformného erytému ( EM) a prípady súvisiace so Stevens-Johnsonovým syndrómom (SJS) a toxickou epidermálnou nekrolýzou (NET), niektoré z nich smrteľné. So známkami alebo príznakmi SJS, NET alebo EM (napr. Kožná vyrážka často s pľuzgiermi alebo slizničnými léziami) liečba sunitinib sa má vysadiť. Ak sa potvrdí diagnóza SJS alebo NET, v liečbe sa nemá pokračovať. V niektorých prípadoch podozrenia na EM po ústupe reakcie pacienti opätovne začínajú liečbu sunitinibom v nižších dávkach tolerovať; niektorí z týchto pacientov tiež dostali súbežná liečba kortikosteroidmi alebo antiseptikmi taminy.
Krvácanie a krvácanie spôsobené nádormi
Po uvedení lieku na trh boli hlásené hemoragické príhody, niektoré s fatálnymi následkami, vrátane gastrointestinálneho, respiračného, močového a mozgového krvácania.
Epizódy krvácania sa vyskytli u 18% pacientov liečených sunitinibom v porovnaní so 17% pacientov v skupine s placebom v štúdii GIST fázy 3. Pacienti s MRCC na sunitinibe, ktorí nikdy predtým neboli liečení sunitinibom, hlásili krvácanie v 39% prípadov v porovnaní s 11%. pacientov liečených IFN-α. Sedemnásť (4,5%) pacientov na sunitinibe v porovnaní s 5 (1,7%) pacientmi na IFN-α malo epizódy krvácania 3. alebo vyššieho stupňa. Dvadsaťšesť percent pacientov, ktorí dostávali sunitinib na cytokíny refraktérnu MRCC, hlásilo epizódy krvácania. Krvácanie, s výnimkou epistaxy, sa vyskytlo u 21,7% pacientov liečených sunitinibom v porovnaní s 9,85% pacientov v skupine s placebom v štúdii fázy 3 pNET. Rutinné vyhodnotenie tejto udalosti by malo zahŕňať kompletný krvný obraz a fyzické vyšetrenie.
Epistaxa bola najčastejšou nežiaducou hemoragickou reakciou, ktorá bola hlásená približne u polovice pacientov so solídnymi nádormi, ktorí hlásili krvácavé príhody. Niektoré z týchto epizód epistaxe boli závažné, ale veľmi zriedkavo smrteľné.
Boli hlásené prípady krvácania z nádoru, niekedy spojené s nekrózou nádoru; niektoré z týchto krvácavých príhod boli smrteľné.
V klinických štúdiách sa „nádorové krvácanie vyskytlo u približne 2% pacientov s GIST. Tieto príhody sa môžu vyskytnúť náhle a v prípade rakoviny pľúc sa môžu prejaviť vo forme„ závažnej a život ohrozujúcej hemoptýzy alebo ako pľúcne krvácanie. V klinických štúdiách boli pozorované prípady pľúcneho krvácania, niektoré s fatálnym koncom, a boli hlásené aj po uvedení lieku na trh u pacientov liečených sunitinibom na liečbu MRCC, GIST a rakoviny pľúc. SUTENT nie je schválený na použitie u pacientov s rakovinou pľúc.
Pacienti súbežne liečení antikoagulanciami (napr.warfarín, acenokumarol) je možné pravidelne monitorovať pomocou kompletného krvného obrazu (krvných doštičiek), koagulačných faktorov (PT / INR) a fyzického vyšetrenia.
Poruchy gastrointestinálneho traktu
Hnačka, nauzea / vracanie, bolesť brucha, dyspepsia a stomatitída / bolesť úst boli najčastejšie hlásenými gastrointestinálnymi nežiaducimi reakciami; Boli hlásené aj prípady ezofagitídy (pozri časť 4.8).
Podporná starostlivosť o gastrointestinálne nežiaduce reakcie vyžadujúce liečbu môže zahŕňať lieky s antiemetickými, antidiarrheálnymi alebo antacidovými vlastnosťami.
U pacientov s intraabdominálnou malignitou liečených sunitinibom sa vyskytli závažné, niekedy smrteľné, gastrointestinálne komplikácie, vrátane gastrointestinálnej perforácie. V štúdii GIST fázy 3 sa fatálne gastrointestinálne krvácanie vyskytlo u 0,98% pacientov liečených placebom.
Hypertenzia
Hypertenzia bola veľmi častou nežiaducou reakciou hlásenou v klinických štúdiách. Dávka sunitinibu bola znížená alebo jeho podanie dočasne pozastavené u približne 2,7% pacientov, u ktorých sa vyskytla hypertenzia. U žiadneho z týchto pacientov nebol sunitinib natrvalo vysadený. Ťažká hypertenzia (> 200 mmHg systolický alebo 110 mmHg diastolický) sa vyskytol u 4,7% pacientov so solídnymi nádormi. U MRCC a predtým neliečených pacientov boli prípady hypertenzie hlásené u 33,9% pacientov na sunitinibe v porovnaní s 3,6% pacientov na IFN-α. závažná hypertenzia sa vyskytla u 12% predtým neliečených pacientov v skupine so sunitinibom a u menej ako 1% pacientov v skupine s IFN-α. V štúdii fázy 3 pNET bola hypertenzia hlásená u 26,5% pacientov užívajúcich sunitinib v porovnaní so 4,9% pacientov v skupine s placebom. Epizódy ťažkej hypertenzie sa vyskytli u pacientov s pNET u 10% pacientov liečených sunitinibom a 3% pacientov Tí, ktorí sú liečení placebom, majú byť vyšetrení na hypertenziu a vhodne monitorovaní. U pacientov s ťažkou nekontrolovanou hypertenziou s medikamentóznou liečbou sa odporúča dočasné prerušenie liečby. Liečba môže pokračovať, ak je hypertenzia dostatočne kontrolovaná.
Hematologické poruchy
Absolútne zníženie počtu neutrofilov 3. a 4. stupňa bolo pozorované u 10% a 1,7% pacientov zaradených do štúdie GIST fázy 3 a u 16% a 1,6% pacientov zaradených do štúdie. Fáza 3 MRCC a u 13 % a 2,4% pacientov zaradených do štúdie pNET fázy 3. Zníženie počtu krvných doštičiek 3. a 4. stupňa bolo hlásené u 3,7%, respektíve 0,4% pacientov. pacientov zaradených do štúdie fázy 3 GIST, u 8,2% a 1,1% pacientov zaradených do štúdie fázy 3 MRCC a u 3,7% a 1,2% pacientov zaradených do štúdie fázy 3 na pNET.
Vyššie uvedené udalosti neboli kumulatívne, boli spravidla reverzibilné a zvyčajne neviedli k prerušeniu liečby. Žiadna z týchto udalostí v štúdiách fázy 3 nebola smrteľná, ale v postmarketingovej fáze lieku neboli hlásené zriedkavé hematologické príhody. vrátane krvácania spojeného s trombocytopéniou a neutropenickými infekciami.
Pozorovala sa anémia v počiatočných aj neskorých štádiách liečby sunitinibom; boli hlásené 3. a 4. stupeň.
U pacientov užívajúcich sunitinib sa má na začiatku každého liečebného cyklu vykonať kompletný krvný obraz.
Srdcové patológie
U pacientov liečených sunitinibom boli hlásené kardiovaskulárne príhody, niektoré z nich smrteľné, vrátane srdcového zlyhania, kardiomyopatie, ischémie myokardu a infarktu myokardu. Tieto údaje naznačujú, že sunitinib zvyšuje riziko kardiomyopatie. U liečených pacientov neboli identifikované žiadne ďalšie špecifické rizikové faktory pre kardiomyopatiu indukovanú sunitinibom okrem špecifického účinku lieku. Sunitinib sa má používať s opatrnosťou u pacientov s rizikom týchto príhod alebo u ktorých sa tieto príhody vyskytli v anamnéze.
V klinických štúdiách došlo k zníženiu ejekčnej frakcie ľavej komory (LVEF) o ≥ 20% a pod dolnú hranicu normálu približne u 2% pacientov s GIST liečených sunitinibom, u 4% pacientov s MRCC refraktérnymi na cytokíny liečených sunitinibom a u 2 % placebom liečených pacientov s GIST. Tieto poklesy LVEF sa nezdajú byť progresívne a často sa zlepšujú s pokračujúcou liečbou. V štúdii vykonanej u pacientov s MRCC a nikdy predtým neliečených malo 27% pacientov liečených sunitinibom a 15% pacientov liečených IFN-α hodnotu LVEF pod dolnou hranicou normálu. Dvom pacientom (
U pacientov s GIST boli epizódy „srdcového zlyhania“, „kongestívneho srdcového zlyhania“ alebo „zlyhania ľavej komory“ hlásené u 1,2% pacientov liečených sunitinibom a u 1% pacientov liečených placebom. V kľúčovej štúdii vo fáze 3 na GIST (n = 312), fatálne srdcové reakcie súvisiace s liečbou sa vyskytli u 1% pacientov v oboch ramenách štúdie (rameno sunitinibu a placeba). V štúdii fázy 2 u pacientov s cytokínmi refraktérnym MRCC hlásilo 0,9 % pacientov fatálny infarkt myokardu súvisiaci s liečbou a v štúdii fázy 3 s pacientmi s MRCC a nikdy predtým neliečenými 0,6 % pacientov v ramene IFN-α a 0% pacientov v ramene sunitinibu hlásilo smrteľné srdcové príhody. V štúdii fázy 3 pNET mal jeden pacient (1%) užívajúci sunitinib smrteľné srdcové zlyhanie súvisiace s liečbou. Možná korelácia medzi inhibíciou receptora tyrozínkinázy (RTK) a srdcovou funkciou nie je jasná.
Pacienti, ktorí do 12 mesiacov pred podaním sunitinibu hlásili srdcové príhody, ako je infarkt myokardu (vrátane ťažkej / nestabilnej angíny), koronárny / periférny bypas, symptomatický CHF, cerebrovaskulárna príhoda alebo prechodný ischemický záchvat alebo pľúcna embólia, boli vylúčení z klinické štúdie so sunitinibom. Nie je známe, či môžu mať pacienti s takýmito sprievodnými stavmi zvýšené riziko vzniku dysfunkcie ľavej komory súvisiacej s liekom.
Zvlášť u pacientov so srdcovými rizikovými faktormi a / alebo s anamnézou ischemickej choroby srdca sa má starostlivo sledovať výskyt klinických príznakov a symptómov kongestívneho srdcového zlyhania.
Lekárom sa odporúča, aby zvážili toto riziko oproti potenciálnym prínosom lieku. Títo pacienti majú byť počas liečby sunitinibom starostlivo sledovaní kvôli klinickým príznakom a symptómom kongestívneho srdcového zlyhania. Keď je pacient liečený sunitinibom, má sa zvážiť aj východiskové a periodické hodnotenie ejekčnej frakcie ľavej komory. U pacientov bez srdcových rizikových faktorov by sa malo zvážiť zhodnotenie ventrikulárnej ejekčnej frakcie na začiatku.
V prítomnosti klinických prejavov CHF sa odporúča prerušiť liečbu sunitinibom. Podávanie sunitinibu sa má prerušiť a / alebo znížiť dávka u pacientov bez klinického prejavu kongestívneho srdcového zlyhania, ale s poklesom ejekčnej frakcie medzi 20% a 50%. od základnej línie.
Predĺženie QT intervalu
Údaje z predklinických štúdií (in vitro A in vivo), ktoré sa vykonávajú s dávkami vyššími ako sú odporúčané pre ľudí, naznačujú, že sunitinib môže inhibovať procesy srdcovej repolarizácie (napr. predĺženie QT intervalu).
Zvýšenie intervalu QTc na viac ako 500 ms sa vyskytlo rýchlosťou 0,5% a zmeny oproti východiskovým hodnotám o viac ako 60 ms sa vyskytli u 1,1% zo 450 pacientov so solídnymi nádormi; oba tieto parametre sú považované za potenciálne významné variácie. Pri koncentráciách zodpovedajúcich približne dvojnásobku terapeutických koncentrácií sa zistilo, že sunitinib predlžuje interval QTcF (korigované podľa Fredericovho vzorca).
Predĺženie QT intervalu bolo študované v štúdii zahŕňajúcej 24 pacientov vo veku 20-87 rokov s pokročilými malignitami. Výsledky tejto štúdie ukázali, že sunitinib má vplyv na QTc interval (definovaný ako priemerná zmena upravená o placebo> 10 ms, s horná hranica 90% CI> 15 ms) pri terapeutických koncentráciách (deň 3) s použitím 24-hodinovej základnej korekčnej metódy a pri koncentráciách nad terapeutickými koncentráciami (deň 9) pri použití oboch korekčných metód na začiatku. Žiadny pacient neoznámil hodnotu QTc> 500 ms.Aj keď bol účinok na interval QTcF pozorovaný na 3. deň 24 hodín po podaní dávky (t.j. s očakávanou terapeutickou plazmatickou koncentráciou po odporúčanej počiatočnej dávke 50 mg) s 24-hodinovou základnou korekčnou metódou, klinický význam tohto zistenia nie je jasný. .
Použitím komplexných sériových hodnotení EKG zodpovedajúcich terapeutickým expozíciám alebo expozíciám nad terapeutickým hodnotám nebol u žiadneho z pacientov v hodnotiteľných populáciách alebo ITT populáciách predĺžený „závažný“ QTc interval (teda rovnaký alebo väčší ako
3. stupeň CTCAE verzie 3.0).
Pri terapeutických plazmatických koncentráciách bola priemerná maximálna zmena v intervale QTcF (korigovaná Fredericiným vzorcom) oproti východiskovým hodnotám 9,6 ms (90% IS 15,1 ms). Pri terapeutických koncentráciách zodpovedajúcich približne dvojnásobku terapeutických koncentrácií bola maximálna zmena intervalu QTcF oproti východiskovým hodnotám bolo 15,4 ms (90% CI 22,4 ms).
Moxifloxacín (400 mg) použitý ako pozitívna kontrola vykazoval priemernú maximálnu zmenu intervalu QTcF oproti východiskovým hodnotám 5,6 ms. Žiadny subjekt neoznámil účinok na interval QTc vyšší ako stupeň 2 (CTCAE verzia 3.0).
Predĺženie QT intervalu môže spôsobiť zvýšené riziko ventrikulárnych arytmií vrátane Torsade de Pointes. Torsade de Pointes sa pozorovalo v
Sunitinib sa má používať opatrne u pacientov s anamnézou predĺženia QT intervalu, u pacientov liečených antiarytmikami alebo liekmi, ktoré môžu predĺžiť QT interval, alebo u pacientov s už existujúcim závažným srdcovým ochorením, bradykardiou alebo abnormalitami. Súbežné podávanie sunitinibu so silnými inhibítormi CYP3A4 má byť obmedzené z dôvodu možného zvýšenia plazmatických koncentrácií sunitinibu (pozri časti 4.2 a 4.5).
Venózne tromboembolické príhody
Venózne tromboembolické príhody súvisiace s liečbou boli pozorované u približne 1,0% pacientov so solídnymi nádormi liečených sunitinibom v klinických štúdiách vrátane štúdií GIST a MRCC.
V štúdii GIST fázy 3 zažilo sedem pacientov (3%), ktorí dostávali sunitinib, a žiadni pacienti v skupine s placebom, venózne tromboembolické príhody; päť zo siedmich pacientov malo hlbokú žilovú trombózu 3. stupňa (DVT) a dvaja mali hlbokú žilovú trombózu stupňa 1 alebo 2. Štyria zo siedmich pacientov liečených na GIST boli po pozorovaní DVT prerušení.
Trinásť pacientov (3%) liečených sunitinibom v štúdii fázy 3 MRCC a nikdy predtým neliečených a štyria pacienti (2%) z dvoch štúdií MRCC rezistentných na cytokíny hlásili venózne tromboembolické príhody. Deväť z týchto pacientov malo venózne tromboembolické príhody. “ pľúcna embólia, jeden so stupňom 2 a osem so stupňom 4. Osem z týchto pacientov malo DVT, jeden so stupňom 1, dvaja so stupňom 2, štyria so stupňom 3 a jeden so stupňom 4. U pacienta s pľúcnou embóliou v štúdii MRCC , odolná voči cytokínom, dávka bola zastavená.
Z predtým neliečených pacientov s MRCC na IFN-α šesť (2%) hlásilo venózne tromboembolické príhody, jeden pacient (pľúcna embólia, všetky 4. stupňa).
V štúdii pNET fázy 3 boli venózne tromboembolické príhody hlásené u 1 (1,2%) pacienta v ramene so sunitinibom a u 5 (6,1%) pacientov v ramene s placebom. Dva z týchto subjektov liečených placebom hlásili DVT, jeden stupeň 2 a jeden stupeň 3.
V pivotných štúdiách GIST, MRCC a pNET neboli pozorované žiadne prípady s fatálnym koncom. Po uvedení lieku na trh boli pozorované prípady s fatálnymi následkami (pozri respiračné príhody a časť 4.8).
Arteriálne tromboembolické príhody
U pacientov liečených sunitinibom boli hlásené prípady arteriálnych tromboembolických príhod (ATE), niekedy smrteľné. K najčastejším príhodám patrili cievna mozgová príhoda, prechodný ischemický záchvat a mozgová ischémia. Rizikové faktory spojené s arteriálnymi tromboembolickými príhodami, okrem už existujúcej malignity a veku 65 rokov a starších, zahŕňali hypertenziu, diabetes mellitus a predchádzajúcu tromboembolickú príhodu.
Respiračné príhody
Pacienti, ktorí mali pľúcnu embóliu počas predchádzajúcich 12 mesiacov, boli vylúčení z klinických štúdií so sunitinibom.
U pacientov, ktorí dostávali sunitinib v pivotných štúdiách fázy 3, boli pľúcne príhody (dyspnoe, pleurálny výpotok, pľúcna embólia alebo pľúcny edém) pozorované u približne 17,8% pacientov s GIST, u približne 26,7% pacientov s MRCC a u 12% pacienti s pNET.
Približne 22,2% pacientov so solídnymi nádormi vrátane GIST a MRCC liečených sunitinibom v klinických štúdiách hlásilo pľúcne príhody.
Prípady pľúcnej embólie boli pozorované u približne 3,1% pacientov s GIST a približne 1,2% pacientov s MRCC liečených sunitinibom v štúdiách fázy 3 (pozri časť 4.4. - Venózne tromboembolické príhody). U pacientov s pNET sa nepozorovali žiadne prípady pľúcnej embólie liečení sunitinibom v štúdii fázy 3. Po uvedení lieku na trh boli pozorované zriedkavé prípady s fatálnym koncom (pozri časť 4.8).
Dysfunkcia štítnej žľazy
Odporúča sa vyhodnotiť funkciu štítnej žľazy meraním východiskových laboratórnych hodnôt u všetkých pacientov. Pacienti s už existujúcou hypotyreózou alebo hypertyreózou by mali byť pred začatím liečby sunitinibom liečení podľa štandardnej klinickej praxe. Počas liečby sunitinibom by sa mala vykonávať pravidelná kontrola funkcie štítnej žľazy každé 3 mesiace. Okrem toho by mali byť všetci pacienti počas liečby starostlivo sledovaní kvôli možným prejavom a symptómom dysfunkcie štítnej žľazy a pacienti, u ktorých sa objavia akékoľvek znaky a / alebo symptómy naznačujúce dysfunkciu štítnej žľazy, by mali podstúpiť laboratórne vyšetrenie funkcie štítnej žľazy, ako sa klinicky očakáva. Pacienti, u ktorých sa vyvinie dysfunkcia štítnej žľazy, by mali byť liečení podľa štandardnej klinickej praxe.
Hypotyreóza sa pozorovala na začiatku alebo na konci liečby sunitinibom.
Hypotyreóza bola hlásená ako nežiaduca reakcia u 7 pacientov (4%), ktorí dostávali sunitinib v dvoch štúdiách MRCC vykonaných u pacientov odolných voči cytokínom; u 61 pacientov (16%), ktorí dostávali sunitinib a u troch pacientov (
Okrem toho bolo hlásené zvýšenie TSH u 4 pacientov (2%) liečených na cytokíny refraktérny MRCC. Celkovo 7% populácie MRCC uviedlo klinický alebo laboratórny dôkaz hypotyreózy súvisiacej s liečbou. Získaná hypotyreóza bola pozorovaná u 6,2% pacientov v štúdii GIST užívajúcich sunitinib v porovnaní s 1% v skupine s placebom. V štúdii fázy 3 pNET bola hypotyreóza hlásená u 6 pacientov (7,2%), ktorí dostávali sunitinib a u jedného pacienta (1,2%), ktorí dostávali placebo.
Funkcia štítnej žľazy bola prospektívne monitorovaná v dvoch štúdiách u pacientok s rakovinou prsníka; SUTENT nie je schválený na liečbu rakoviny prsníka. V jednej štúdii bola hypotyreóza hlásená u 15 subjektov (13,6%) liečených sunitinibom a u 3 subjektov (2,9%) liečených štandardnou liečbou. Zvýšený TSH v krvi bol hlásený u 1 subjektu. (0,9%) liečených sunitinibom a u žiadnych subjektov liečené štandardnou terapiou. Hypertyreóza nebola hlásená u žiadneho zo subjektov liečených sunitinibom a bola hlásená u 1 subjektu (1,0%), ktorý dostával štandardnú liečbu. V druhej štúdii bola hypotyreóza hlásená u celkom 31 subjektov (13%) liečených sunitinibom a 2 subjektov (0,8%) liečených kapecitabínom. Zvýšenie TSH v krvi bolo hlásené u 12 subjektov (5,0%) liečených sunitinibom a u žiadnych subjektov liečených kapecitabínom. Hypertyreóza bola hlásená u 4 subjektov (1,7%) liečených sunitinibom a u žiadnych subjektov liečených kapecitabínom. Zníženie krvného TSH bolo hlásené u 3 subjektov (1,3%) liečených sunitinibom a u žiadnych subjektov. Liečených kapecitabínom. Zvýšenie T4 bolo hlásené u 2 subjekty (0,8%) liečené sunitinibom a u 1 subjektu (0,4%) liečené kapecitabínom. Zvýšenie T3 bolo hlásené u 1 subjektu (0,8%) liečeného sunitinibom a u žiadneho subjektu liečeného kapecitabínom.Všetky hlásené udalosti súvisiace so štítnou žľazou boli 1. až 2. stupňa.
V klinických skúšaniach a vo fáze uvádzania lieku na trh boli menej často hlásené prípady hypertyreózy, niektoré nasledované hypotyreózou a prípady tyreoiditídy.
Pankreatitída
Zvýšenie aktivity sérovej lipázy a amylázy bolo pozorované u pacientov s niekoľkými solídnymi nádormi, ktorí dostávali sunitinib. Zvýšenie aktivity sérovej lipázy bolo prechodné a spravidla nesúviselo so znakmi a symptómami pankreatitídy u subjektov so solídnymi nádormi rôznych typov.
Pankreatitída bola pozorovaná menej často (
Boli hlásené prípady závažných pankreatických príhod, niektoré s fatálnym koncom.
Ak sa objavia príznaky pankreatitídy, liečba sunitinibom sa má prerušiť a pacientom by mala byť poskytnutá primeraná podporná starostlivosť. V štúdii fázy 3 pNET neboli hlásené žiadne prípady pankreatitídy súvisiace s liečbou.
Hepatotoxicita
U pacientov liečených sunitinibom bola pozorovaná hepatotoxicita. Pri funkcii pečene (alanín transamináza [ALT], aspartát transamináza [AST], hladiny bilirubínu) boli pozorované prípady zlyhania pečene, niekedy s fatálnym koncom, prejavy alebo príznaky zlyhania pečene, liečbu sunitinibom je potrebné prerušiť a pacientom by mala byť poskytnutá primeraná podporná lekárska starostlivosť.
Poruchy pečene a žlčových ciest
Liečba sunitinibom môže byť spojená s cholecystitídou vrátane alitázickej cholecystitídy a enfematóznej cholecystitídy. V kľúčových klinických štúdiách bol výskyt cholecystitídy 0,5%.
Po uvedení lieku na trh boli hlásené prípady cholecystitídy.
Funkcia obličiek
Boli hlásené prípady poškodenia funkcie obličiek, zlyhania obličiek a / alebo akútneho zlyhania obličiek, v niektorých prípadoch s fatálnym koncom.
Medzi rizikové faktory spojené s poruchou / zlyhaním obličiek u pacientov užívajúcich sunitinib patrí okrem už existujúceho karcinómu obličkových buniek: pokročilý vek, diabetes mellitus, už existujúce poškodenie funkcie obličiek, srdcové zlyhanie, hypertenzia, sepsa, dehydratácia / hypovolémia a rabdomyolýza.
Bezpečnosť pokračovania liečby sunitinibom u pacientov so stredne ťažkou až ťažkou proteinúriou nebola systematicky hodnotená.
Boli hlásené prípady proteinúrie a zriedkavé prípady nefrotického syndrómu. Odporúča sa východisková analýza moču a pacienti by mali byť sledovaní z hľadiska možného vývoja alebo zhoršenia proteinúrie.
U pacientov s nefrotickým syndrómom sa má liečba sunitinibom prerušiť.
Fistuly
Ak dôjde k tvorbe fistuly, liečba sunitinibom sa má prerušiť. K dispozícii sú obmedzené informácie o predĺženej liečbe sunitinibom u pacientov s fistulami.
Porušenie procesu hojenia rán
Počas liečby sunitinibom boli hlásené prípady zhoršeného hojenia rán. Neuskutočnili sa žiadne formálne klinické štúdie o účinku sunitinibu na hojenie rán. Z preventívnych dôvodov sa odporúča dočasne prerušiť liečbu sunitinibom u pacientov podstupujúcich veľký chirurgický zákrok. Klinické skúsenosti týkajúce sa načasovania potrebného na opätovné spustenie. Terapia po veľkom chirurgickom zákroku je obmedzená. Preto rozhodnutie obnoviť liečbu sunitinibom po veľkom chirurgickom zákroku musí byť založené na klinickom posúdení zotavenia z chirurgického zákroku.
Osteonekróza dolnej / dolnej čeľuste
U pacientov liečených SUTENTOM boli hlásené prípady osteonekrózy čeľuste. Vo väčšine hlásených prípadov boli pacienti predtým alebo súčasne liečení intravenóznymi bisfosfonátmi, u ktorých je identifikovaným rizikom osteonekróza čeľuste. Preto je potrebná opatrnosť, ak sa SUTENT a intravenózne bisfosfonáty podávajú súbežne alebo kombinovane.
Invazívne zubné intervencie boli tiež identifikované ako rizikový faktor. Pred liečbou SUTENTom je potrebné zvážiť zubné vyšetrenie a vhodnú preventívnu zubnú starostlivosť. Pokiaľ je to možné, je potrebné vyhnúť sa invazívnym zubným zákrokom u pacientov, ktorí predtým užívali alebo užívajú intravenózne bisfosfonáty (pozri časť 4.8).
Precitlivenosť / angioedém
Ak dôjde k edému v dôsledku reakcie z precitlivenosti, liečba sunitinibom sa má prerušiť a začať štandardná lekárska liečba.
Poruchy nervového systému
Poruchy chuti
Dysgeúzia bola hlásená u približne 28% pacientov užívajúcich sunitinib počas klinických štúdií.
Kŕče
V klinických štúdiách so sunitinibom a po uvedení lieku na trh boli u subjektov s rádiologickými dôkazmi mozgových metastáz alebo bez nich pozorované prípady záchvatov. Okrem toho existuje obmedzený počet hlásení (bolesť hlavy, znížená ostražitosť, zhoršené mentálne funkcie a strata zraku vrátane kortikálnej slepoty by mali byť kontrolované liečbou vrátane kontroly hypertenzie. Odporúča sa dočasné pozastavenie liečby sunitinibom; po úprave v takom prípade môže byť liečba obnovená podľa uváženia ošetrujúceho lekára.
Syndróm nádorovej lýzy (TLS)
Prípady syndrómu rozpadu nádoru (TLS), niektoré s fatálnym koncom, boli v klinických štúdiách zriedkavo pozorované a boli hlásené po uvedení na trh u pacientov liečených sunitinibom. Medzi rizikové faktory TLS patrí vysoká nádorová záťaž, už existujúce chronické zlyhanie obličiek, oligúria, dehydratácia, hypotenzia a kyslý moč. Títo pacienti by mali byť starostlivo monitorovaní a liečení tak, ako je to klinicky indikované; u týchto pacientov treba zvážiť profylaktickú hydratáciu.
Infekcie
Boli hlásené prípady závažných infekcií s neutropéniou alebo bez neutropénie, vrátane niektorých prípadov s fatálnym koncom.
Infekcie, ktoré sa najčastejšie vyskytujú pri liečbe sunitinibom, sú infekcie typické pre onkologických pacientov, ako sú dýchacie cesty, močové cesty, koža a sepsa.
Boli hlásené zriedkavé, niekedy smrteľné prípady nekrotizujúcej fasciitídy vrátane perinea. Liečba sunitinibom sa má ukončiť u pacientov, u ktorých sa vyvinie nekrotizujúca fasciitída, a ihneď začať vhodnú liečbu.
Hypoglykémia
Počas liečby sunitinibom boli hlásené znížené glykémie, niektoré z nich klinicky symptomatické a vyžadujúce hospitalizáciu z dôvodu straty vedomia. V prípade symptomatickej hypoglykémie sa má liečba sunitinibom dočasne prerušiť. U diabetických pacientov by sa mala pravidelne kontrolovať hladina glukózy v krvi, aby sa posúdilo, či je potrebné upraviť dávkovanie liekov na diabetes, aby sa minimalizovalo riziko hypoglykémie.
04.5 Interakcie s inými liekmi a iné formy interakcie
Interakčné štúdie boli vykonané len u dospelých.
Lieky, ktoré môžu zvýšiť plazmatické koncentrácie sunitinibu
U zdravých dobrovoľníkov malo súbežné podanie jednorazovej dávky sunitinibu s ketokonazolom, silným inhibítorom CYP3A4, za následok zvýšenie kombinovanej Cmax [sunitinib + primárny metabolit] o 49% a AUC0-∞ o 51%.
Podávanie sunitinibu so silnými inhibítormi CYP3A4 (napr. Ritonavirom , itrakonazol, erytromycín, klaritromycín, grapefruitový džús) môžu zvýšiť koncentrácie sunitinibu.
Preto sa treba vyhnúť kombinácii s inhibítormi CYP3A4 alebo zvážiť alternatívny liek, ktorý nemá žiadny alebo má minimálny potenciál inhibovať CYP3A4.
Ak to nie je možné, dávku SUTENTU možno bude potrebné znížiť na minimum 37,5 mg / deň pre GIST a MRCC alebo 25 mg / deň pre pNET na základe starostlivého monitorovania znášanlivosti (pozri časť 4.2).
Lieky, ktoré môžu redukovať plazmatické koncentrácie sunitinibu
U zdravých dobrovoľníkov malo súbežné podanie jednej dávky sunitinibu a rifampicínu induktora CYP3A4 za následok zníženie kombinovanej Cmax [sunitinib + primárny metabolit] o 23% a AUC0-∞ o 46%.
Podávanie sunitinibu so silnými induktormi CYP3A4 (napr. Dexametazón, fenytoín, karbamazepín, rifampicín, fenobarbital alebo bylinné prípravky s obsahom ľubovníka bodkovaného /Hypericum perforatum) môže znížiť koncentrácie sunitinibu. Je preto potrebné vyhnúť sa asociácii s induktormi CYP3A4 alebo je potrebné zvážiť alternatívny liek, ktorý nemá žiadny alebo má minimálny potenciál indukovať CYP3A4. Ak to nie je možné, dávku SUTENTU je možné zvyšovať v prírastkoch 12, 5 mg (až do na 87,5 mg / deň pre GIST a MRCC alebo 62,5 mg / deň pre pNET) na základe starostlivého monitorovania znášanlivosti (pozri časť 4.2).
04.6 Gravidita a laktácia
Tehotenstvo
Neuskutočnili sa žiadne štúdie u gravidných žien užívajúcich sunitinib. Štúdie na zvieratách preukázali reprodukčnú toxicitu vrátane malformácií plodu (pozri časť 5.3).
SUTENT sa nemá používať počas tehotenstva alebo u žien, ktoré nepoužívajú účinné antikoncepčné opatrenia, pokiaľ potenciálne prínosy neodôvodňujú potenciálne riziko pre plod. Ak sa SUTENT používa počas gravidity alebo ak pacientka otehotnie počas liečby SUTENTOM, musí byť pacientka informovaná o potenciálnych rizikách pre plod.
Ženám vo fertilnom veku treba odporučiť, aby používali účinnú antikoncepciu a aby sa počas liečby SUTENTOM vyhli otehotneniu.
Čas kŕmenia
Sunitinib a / alebo jeho metabolity sa vylučujú do mlieka potkanov. Nie je známe, či sa sunitinib alebo jeho hlavný aktívny metabolit vylučujú do ľudského mlieka. Pretože sa účinné látky vo všeobecnosti vylučujú do materského mlieka a vzhľadom na možné závažné nežiaduce reakcie u dojčených detí, ženy nemajú počas liečby SUTENTom dojčiť.
Plodnosť
Predklinické údaje naznačujú, že fertilita mužov a žien môže byť pri liečbe sunitinibom narušená (pozri časť 5.3).
04.7 Účinky na schopnosť viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje.Pacienti majú byť informovaní o možnom výskyte závratov počas liečby sunitinibom.
04.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Najzávažnejšie nežiaduce reakcie súvisiace s liečbou sunitinibom, niektoré smrteľné, sú zlyhanie obličiek, srdcové zlyhanie, pľúcna embólia, gastrointestinálna perforácia a krvácanie (napr. Respiračné, gastrointestinálne, s nádorom súvisiace, močové a mozgové krvácanie).
Medzi najčastejšie nežiaduce reakcie akéhokoľvek stupňa (hlásené pacientmi v pivotných klinických štúdiách s RCC, GIST a pNET) patrí znížená chuť do jedla, poruchy chuti, hypertenzia, únava, gastrointestinálne poruchy (napr. Hnačka, nauzea, stomatitída, dyspepsia a vracanie), zmena farby syndrómu kože a palmárno-plantárnej erytrodysestézie. Tieto príznaky sa môžu pri pokračujúcej liečbe zmierniť. Počas liečby môže dôjsť k hypotyreóze.Hematologické poruchy (napr. Neutropénia, trombocytopénia a anémia) patrili k najčastejším nežiaducim reakciám na liek.
Smrteľné nežiaduce udalosti iné ako tie, ktoré sú uvedené v časti 4.4. alebo v časti 4.8 a považované za pravdepodobne súvisiace so sunitinibom zahŕňajú zlyhanie viacerých orgánov, diseminovanú intravaskulárnu koaguláciu, peritoneálne krvácanie, adrenálnu insuficienciu, pneumotorax, šok a náhlu smrť.
Tabuľka nežiaducich reakcií
Nežiaduce reakcie hlásené pacientmi s GIST, MRCC a pNET v súbore údajov 7115 pacientov sú uvedené nižšie a kategorizované podľa triedy orgánových systémov, frekvencie a závažnosti (NCI-CTCAE). Hlásené sú tiež postmarketingové nežiaduce reakcie identifikované v klinických štúdiách. V rámci každej frekvenčnej triedy sú nežiaduce účinky uvedené v poradí podľa klesajúcej závažnosti.
Frekvencie sú definované nasledovne: veľmi časté (≥1 / 10), časté (≥1 / 100a
Tabuľka 1 - Nežiaduce reakcie hlásené v klinických štúdiách
Nasledujúce výrazy boli zoskupené:
nazofaryngitída a orálny herpes
b Bronchitída, infekcia dolných dýchacích ciest, zápal pľúc a infekcia dýchacích ciest
c Absces, končatinový absces, análny absces, gumový absces, pečeňový absces, pankreatický absces, perineálny absces, perirektálny absces, rektálny absces, subkutánny absces a zubný absces
d Kandidóza pažeráka a orálna kandidóza
a celulitída a infekcia kože
f Sepsa a septický šok
g Abdominálny absces, abdominálna sepsa, divertikulitída a osteomyelitída
h Znížená chuť do jedla a anorexia
i Dysgeúzia, starnutie a zmeny chuti
j Akútny koronárny syndróm, angina pectoris, nestabilná angína, oklúzia koronárnej artérie, ischémia myokardu
k Redukcia / anomália ejekčnej frakcie
l Akútny infarkt myokardu, infarkt myokardu, tichý infarkt myokardu
m Orofaryngeálna a faryngolaryngeálna bolesť
n Stomatitída a aftózna stomatitída
o Bolesť brucha, dolná a horná časť brucha
p Gastrointestinálna a intestinálna perforácia
q Cholecystitída a alitická cholecystitída
r Žltá koža, zmeny farby kože a pigmentácie
s psoriasiformná dermatitída, exfoliatívna vyrážka, vyrážka, erytematózna vyrážka, folikulárna vyrážka, generalizovaná vyrážka, makulárna vyrážka, makulopapulárna vyrážka, papulárna vyrážka a svrbivá vyrážka
t Kožná reakcia a kožná patológia
u Patológia nechtov a zmena farby nechtov
v Únava a asténia
w Edém tváre, edém a periférny edém
x Amyláza a zvýšená amyláza
* Zahŕňa smrteľné udalosti
Popis identifikovaných nežiaducich reakcií
Infekcie a nákazy: Boli hlásené prípady závažných infekcií (s neutropéniou alebo bez neutropénie), vrátane prípadov s fatálnym koncom. Boli hlásené prípady nekrotizujúcej fasciitídy, niekedy smrteľné, ktoré by mohli postihnúť aj oblasť perinea (pozri tiež časť 4.4).
Poruchy krvi a lymfatického systému: Boli hlásené prípady trombotickej mikroangiopatie. V týchto prípadoch sa odporúča dočasné pozastavenie činnosti SUTENT; po vyriešení týchto udalostí možno liečbu znovu začať podľa uváženia ošetrujúceho lekára.
Poruchy imunitného systému: Boli hlásené reakcie z precitlivenosti vrátane angioedému.
Poruchy nervového systému: Bolo hlásených niekoľko prípadov, niektoré smrteľných, subjektov so záchvatmi a rádiologickými dôkazmi o syndróme reverzibilnej zadnej leukoencefalopatie (RPLS) (pozri tiež časť 4.4).
Poruchy metabolizmu a výživy: U pacientov s pNET bol hlásený vyšší výskyt hypoglykemických príhod v porovnaní s pacientmi s MRCC a GIST. Mnohé z nežiaducich udalostí pozorovaných v klinických štúdiách sa však nepovažovali za súvisiace so študijnou liečbou.
Poruchy pečene a žlčových ciest: Bola hlásená hepatálna dysfunkcia, ktorá môže zahŕňať abnormality testov funkcie pečene, hepatitídu alebo zlyhanie pečene.
Poruchy kože a podkožného tkaniva: Boli hlásené prípady gangrenóznej pyodermie, zvyčajne reverzibilné po prerušení liečby (pozri tiež časť 4.4).
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva: Boli hlásené prípady myopatie a / alebo rabdomyolýzy, niektoré súvisiace s akútnym zlyhaním obličiek. Pacienti s prejavmi alebo príznakmi svalovej toxicity by mali byť liečení v súlade so štandardnou lekárskou praxou.
Boli hlásené prípady tvorby fistuly, niekedy spojené s nekrózou nádoru a regresiou, v niektorých prípadoch s fatálnym koncom.
U pacientov liečených SUTENTom boli hlásené prípady osteonekrózy čeľuste, z ktorých mnohé sa vyskytli u pacientov, ktorí rozpoznali rizikové faktory pre osteonekrózu čeľuste, najmä expozíciu intravenóznym bisfosfonátmi a / alebo zubné ochorenie v anamnéze vyžadujúce invazívne zubné intervencie (pozri aj časť 4.4).
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité, pretože umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia. V "Príloha V .
04,9 Predávkovanie
Neexistuje žiadne špecifické antidotum na predávkovanie sunitinibom a liečba predávkovania by mala zahŕňať všeobecné podporné opatrenia. Ak je to uvedené, elimináciu neabsorbovanej účinnej látky je možné dosiahnuť zvracaním alebo výplachom žalúdka. Boli hlásené. Prípady predávkovania; v niektorých v týchto prípadoch boli nežiaduce reakcie, ktoré sa vyskytli, v súlade so známym bezpečnostným profilom sunitinibu.
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy.
ATC kód: LO1XE04.
Mechanizmus akcie
Sunitinib inhibuje receptory viacnásobnej tyrozínkinázy (RTK), ktoré sa podieľajú na raste nádoru, neoangiogenéze nádoru a metastatickej progresii rakoviny. Sunitinib bol identifikovaný ako inhibítor receptorov rastových faktorov odvodených od krvných doštičiek (PDGFRα a PDGFRβ), receptorov vaskulárneho endotelového rastového faktora (VEGFR1, VEGFR2 a VEGFR3), receptoru faktora kmeňových buniek (KIT), receptora tyrozínkinázy FLT3 (Fms-like tyrozínkináza 3), receptor CSF-1R (receptor faktora stimulujúceho kolónie (CSF-1R)) a z gliového receptora neutrofického faktora (RET). Hlavný metabolit vykazuje v biochemických a bunkových testoch účinnosť porovnateľnú so sunitinibom.
Klinická účinnosť a bezpečnosť
Bezpečnosť a klinická účinnosť sunitinibu sa skúmala pri liečbe pacientov s GIST rezistentných na imatinib (tj. Tých pacientov, ktorí pokročili počas alebo po liečbe imatinibom) alebo intolerantných na imatinib (tj tí, ktorí mali významnú toxicitu počas liečby imatinibom ktoré bránili pokračovaniu liečby), pri liečbe pacientov s MRCC a pri liečbe pacientov s neoperovateľným pNET.
Účinnosť je založená na čase do progresie nádoru a na zvýšení prežitia u pacientov s GIST, prežívaní bez progresie a miere objektívnej odpovede u predtým neliečených pacientov s MRCC a MRCC refraktérnych na liečbu cytokínmi s MRCC, respektíve z prežívania bez progresie u pacientov s pNET.
Stromálne nádory gastrointestinálneho traktu (GIST)
Počiatočná otvorená stupňujúca sa štúdia bola vykonaná u pacientov s GIST po zlyhaní imatinibu (priemer maximálnej dennej dávky 800 mg) z dôvodu rezistencie alebo intolerancie. Zaradených bolo 97 pacientov s rôznymi dávkami a dávkovacími režimami; 55 pacientov bolo liečených SUTENT 50 mg podľa odporúčaného 4-týždňového a dvojtýždňového harmonogramu vysadenia lieku (schéma 4/2).
V tejto štúdii bol medián TTP času do progresie 34,0 týždňov (95% IS = 22,0-46,0 týždňa).
Randomizovaná, dvojito zaslepená, placebom kontrolovaná fáza 3 štúdie sunitinibu bola vykonaná u pacientov s GIST, ktorí boli intolerantní alebo mali progresiu ochorenia počas alebo po liečbe imatinibom (priemer maximálnej dennej dávky 800 mg). V tejto štúdii bolo 312 pacientov (2: 1) randomizovaných na liečbu 50 mg sunitinibu alebo placeba, perorálne jedenkrát denne podľa schémy 4/2 až do progresie ochorenia alebo do ukončenia štúdie zo „iného dôvodu (207 pacientov dostalo sunitinib a 105 placebo).
Primárnym koncovým ukazovateľom účinnosti štúdie bol TTP, definovaný ako čas od randomizácie do prvej dokumentácie objektívnej progresie nádoru. V čase vopred špecifikovanej predbežnej analýzy bol stredný TTP so sunitinibom 28,9 týždňov (95% IS = 21,3-34,1 týždňa) ), keď ich hodnotil skúšajúci, a 27,3 týždňa (95% IS = 16,0-32,1 týždňa), keď ich hodnotil nezávislý hodnotiaci výbor a bol štatisticky lepší ako 5,1-týždňový TTP získaný s placebom (95% IS = 4,4-10,1 týždňa, p
Nezávislý hodnotiaci výbor. Rozdiel v celkovom prežití bol štatisticky v prospech sunitinibu [pomer nebezpečnosti: 0,491 95% (CI 0,290-0,831)]; riziko smrti bolo 2 -krát vyššie u pacientov v ramene s placebom ako u pacientov v ramene so sunitinibom.
Po predbežnej analýze účinnosti a bezpečnosti bola na odporúčanie nezávislého DSMB štúdia zaslepená a pacientom v ramene s placebom bolo ponúknuté prejsť na otvorenú liečbu sunitinibom.
V otvorenej liečebnej fáze štúdie bolo sunitinibom liečených 255 pacientov, z toho 99 pacientov pôvodne liečených placebom.
Analýza primárnych a sekundárnych koncových ukazovateľov v otvorenej fáze štúdie opätovne potvrdila výsledky získané v čase predbežnej analýzy, ako je uvedené v tabuľke nižšie.
Tabuľka 2 - Súhrn koncových ukazovateľov účinnosti (populácia ITT)
Medián celkového prežitia (OS) v populácii ITT bol 72,7 týždňa v ramene s liečbou sunitinibom a 64,9 týždňa (HR 0,876, 95% IS 0,679 - 1,129, p = 0,306). V tejto analýze skupina s placebom zahŕňala pacientov randomizovaných na placebo a následne prešli na otvorenú liečbu sunitinibom.
Metastatický karcinóm obličkových buniek (MRCC) u predtým neliečených pacientov
Bola vykonaná randomizovaná, multicentrická, medzinárodná štúdia fázy 3 s cieľom vyhodnotiť účinnosť a bezpečnosť sunitinibu v porovnaní s interferónom IFN-α u predtým neliečených pacientov s MRCC. Sedemstopäťdesiat pacientov bolo randomizovaných 1: 1 do liečebných ramien; dostali liečbu sunitinibom v opakované 6-týždňové cykly. Každý cyklus pozostáva zo 4 týždňov s 50 mg denne perorálne, po ktorých nasledujú 2 týždne bez užitia lieku (schéma 4/2) alebo s IFN-α podávaným orálne subkutánne v dávke 3 milióny jednotiek (MU ) prvý týždeň, 6 MU druhý týždeň a od tretieho týždňa ďalej v dávke 9 MU podľa ošetrenia 3 po sebe nasledujúcich dní každý týždeň.
Priemerná dĺžka liečby bola 11,1 mesiaca (rozsah: 0,4 - 46,1) pri liečbe sunitinibom a 4,1 mesiaca (v rozsahu 0,1 - 45,6) pri liečbe IFN -α. Závažné nežiaduce udalosti súvisiace s liečbou (TRSAE) boli hlásené u 23,7% pacientov užívajúcich sunitinib a 6,9% pacientov, ktorí dostávali IFN-α. Miera prerušenia kvôli nežiaducim udalostiam však bola 20% pre sunitinib a 23% pre IFN-α. Prerušenie liečby sa vyskytlo u 202 pacientov (54%) v skupine so sunitinibom a 141 pacientov (39%) v skupine s IFN-α.
K zníženiu dávky došlo u 194 pacientov (52%) liečených sunitinibom a 98 pacientov (27%) liečených IFN-α. Pacienti boli liečení až do progresie ochorenia alebo stiahnutia štúdie. Primárnym koncovým ukazovateľom účinnosti bolo prežitie bez progresie (PFS).
Plánovaná predbežná analýza ukázala štatisticky významnú výhodu pre SUTENT oproti IFN-α. V tejto štúdii bol priemerný PFS pre skupinu so sunitinibom 47,3 týždňa v porovnaní s 22,0 týždňami pre skupinu IFN-α; pomer rizika bol 0,415 (95% IS: 0,320-0,539, p-hodnota
Liečba sunitinibom bola spojená s dlhším prežitím ako liečba IFN-α. Medián celkového prežívania bol 114,6 týždňov v skupine so sunitinibom (95% IS: 100,1 - 142,9 týždňa) a 94,9 týždňa v skupine s IFN -α (95% CI: 77,7 - 117,0 týždňa) s pomer nebezpečnosti 0,821 (95% IS: 0,673-1,001; p = 0,0510 na základe nestratifikovaného log-rank testu).
Prežívanie bez progresie (PFS) a celkové prežitie (OS) pozorované v populácii pacientov, ktorí chcú liečiť (ITT) a určené rádiologickým laboratórnym hodnotením, sú zhrnuté v nasledujúcich tabuľkách:
Súhrn koncových ukazovateľov účinnosti (populácia ITT)
Cytokín-refraktérny metastatický karcinóm obličkových buniek (MRCC)
Štúdia fázy 2 bola vykonaná so SUTENTom u pacientov refraktérnych na predchádzajúcu liečbu cytokínmi liečených interleukínom-2 alebo IFN-α. 63 pacientov dostalo perorálnu úvodnú dávku 50 mg sunitinibu jedenkrát denne počas 4 po sebe nasledujúcich týždňov, po ktorých nasledovala dvojtýždňová prestávka na dokončenie celého 6-týždňového kurzu (schéma liečby 4 /2). Primárnym koncovým ukazovateľom účinnosti bola miera objektívnej odpovede (Objektívny
Rýchlosť odpovede (ORR)) podľa kritérií RECIST (Kritériá hodnotenia reakcie pri solídnych nádoroch).
V tejto štúdii bola miera objektívnej odpovede 36,5% (95% IS 24,7% -49,6%) a medián času do progresie (TTP) bol 37,7 týždňov (95% IS 24,0 -46,4 týždňa).
Otvorená, jednoramenná, multicentrická, potvrdzujúca štúdia na vyhodnotenie účinnosti a bezpečnosti sunitinibu bola vykonaná u pacientov s MRCC refraktérnymi na predchádzajúcu liečbu cytokínmi. Sto šesť pacientov dostalo najmenej jednu dávku 50 sunitinibu. Mg v rámca schémy 4/2.
Primárnym koncovým ukazovateľom účinnosti tejto štúdie bola miera ORR. Sekundárne cieľové ukazovatele zahŕňali TTP, trvanie odpovede (DR) a celkové prežitie (OS).
V tejto štúdii bola ORR 35,8% (95% IS 26,8% -47,5%). DR a medián OS ešte neboli dosiahnuté.
Pankreatické neuroendokrinné nádory (pNET)
Otvorená, multicentrická, podporná štúdia fázy 2 hodnotila účinnosť a bezpečnosť monoterapie sunitinibom v 50 mg denných dávkach v schéme 4/2 [4 týždne liečby, 2 týždne prestávka] u pacientov s neoperovateľným pNET V súbore 66 pacientov pacientov s rakovinou buniek ostrovčekov pankreasu bol primárny koncový ukazovateľ odpovede 17%.
Bola vykonaná pivotná multicentrická, medzinárodná, randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia fázy 3 sunitinibu v monoterapii u pacientov s neoperovateľným pNET.
Pacienti, u ktorých sa požadovala dokumentovaná progresia ochorenia na základe RECIST počas predchádzajúcich 12 mesiacov, boli randomizovaní (1: 1) na podávanie 37,5 mg sunitinibu jedenkrát denne bez plánovanej ochrannej lehoty (n = 86) alebo placeba (n = 85) .
Primárnym koncovým ukazovateľom bolo hodnotenie prežívania bez progresie (PFS) u pacientov užívajúcich sunitinib oproti pacientom, ktorí dostávali placebo. Ďalšími koncovými ukazovateľmi boli OS, percento ORR, výsledky hlásené pacientom) a bezpečnosť.
Z hľadiska demografických charakteristík boli skupiny pacientov liečených sunitinibom a placebom porovnateľné.Okrem toho 49% pacientov liečených sunitinibom a 52% pacientov užívajúcich placebo malo nefunkčné nádory a 92% pacientov v oboch ramenách malo pečeňové metastázy. Štúdia umožnila použitie somatostatínových analógov. 66% pacientov liečených sunitinibom a 72% pacientov liečených placebom bolo predtým liečených systémovou terapiou. Okrem toho 24% pacientov v skupine so sunitinibom a 22% pacientov v placebe skupina dostávala analógy somatostatínu. Klinicky významná výhoda sunitinibu PFS v porovnaní s placebom bola pozorovaná pri hodnotení skúšajúcim. Medián PFS bol 11,4 mesiaca v ramene so sunitinibom v porovnaní s 5, 5 mesiacmi v ramene s placebom [pomer rizika: 0,418 (95% IS 0,263 (0,662), p-hodnota = 0,0001]; podobné výsledky boli pozorované pri hodnotení reakcie nádoru na základe aplikácie RECIST na meranie nádoru vykonanej vyšetrovateľmi na určenie progresie ochorenia, ako je uvedené v tabuľke 3. A pomer nebezpečnosti v prospech sunitinibu sa pozoroval vo všetkých podskupinách hodnotených z hľadiska východiskových charakteristík vrátane analýzy podľa počtu predchádzajúcich systémových terapií. Celkovo 29 pacientov v skupine so sunitinibom a 24 v skupine s placebom nedostalo žiadnu predchádzajúcu systémovú liečbu; u týchto pacientovpomer nebezpečnosti pre PFS to bolo 0,365 (95% IS 0,156, 0,857), p = 0,0156.
Podobne medzi 57 pacientmi v ramene sunitinibu (vrátane 28 s 1 predchádzajúcou systémovou liečbou a 29 s 2 alebo viacerými systémovými terapiami) a 61 pacientov v ramene s placebom (vrátane 25 s 1 predchádzajúcou systémovou liečbou a 36 s 2 alebo viacerými terapiami systémovými ) l "pomer nebezpečnosti pre PFS to bolo 0,456 (95% IS 0,264, 0,787), p = 0,0036.
Analýza citlivosti PFS bola vykonaná, keď bol PFS založený na vyšetrovateľských meraniach nádoru a keď boli všetky subjekty, cenzurované z iných dôvodov, ako je ukončenie štúdie, považované za udalosti PFS. Táto analýza poskytla konzervatívny odhad účinku liečby sunitinibom a potvrdila primárnu analýzu, pričom preukázala „pomer nebezpečnosti 0,507 (95% IS 0,350, 0,733), p = 0,000193. Pivotná štúdia s pankreatickým NET bola predčasne ukončená na základe odporúčania nezávislého výboru pre hodnotenie liečiv a primárny koncový ukazovateľ bol založený na posúdení skúšajúcim: obe podmienky mohli ovplyvniť odhad účinku liečby.
Aby sa vylúčila zaujatosť pri hodnotení PFS vyšetrovateľom, bol vykonaný nezávislý, zaslepený centrálny prehľad diagnostického zobrazovania; toto preskúmanie podporilo hodnotenie skúšajúceho, ako je uvedené v tabuľke 3.
Tabuľka 3 - Výsledky účinnosti zo štúdie fázy 3 pNET
CI = interval spoľahlivosti, HR = pomer rizika, NA = neaplikovateľné, NR = nedosiahnuté
2-stranný nestratifikovaný test log-rank
b Fisherov presný test
Celkový údaj o prežití nebol pri analýze zrejmý. V ramene so sunitinibom došlo k 9 úmrtiam a v ramene s placebom k 21 úmrtiam. Štatisticky významný rozdiel bol pozorovaný v miere objektívnej odpovede sunitinibu v porovnaní s placebom.
V prípade progresie ochorenia boli pacienti informovaní o liečbe, ktorú dostávali, a pacientom užívajúcim placebo bola ponúknutá možnosť zaradiť sa do inej otvorenej rozšírenej štúdie so sunitinibom. Vzhľadom na skoré ukončenie štúdie boli zostávajúci pacienti informovaní o liečbe, ktorú dostávali, a bol im ponúknutý prístup k otvorenej rozšírenej štúdii so sunitinibom. V štúdii bolo sunitinibom liečených celkom 59 pacientov v skupine s placebom. predĺženia.
Výsledky dotazníka o kvalite života Európskej organizácie pre výskum a liečbu rakoviny (EORTC QLQ-C30) ukázali, že celková kvalita života súvisiaca so zdravím a päť funkčných oblastí (fyzická, rolová, kognitívna, emočná a sociálne) boli konzervatívne u pacientov užívajúcich sunitinib v porovnaní s pacientmi, ktorí dostávali placebo, s malým počtom symptomatických vedľajších účinkov.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so SUTENTOM v jednej alebo vo viacerých podskupinách pediatrickej populácie na liečbu GIST (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so SUTENTOM vo všetkých podskupinách pediatrickej populácie na liečbu obličkových a obličkových karcinómov panvy (s vylúčením nefroblastómu, nefroblastomatózy, čistých buniek sarkómu, mezoblastického nefrómu, obličkovej drene karcinóm a rabdoidný nádor obličky) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií so SUTENTOM vo všetkých podskupinách pediatrickej populácie na liečbu gastroenteropankreatických neuroendokrinných nádorov (s vylúčením neuroblastómu, neuroganglioblastómu, feochromocytómu) (informácie o použití v pediatrickej populácii, pozri časť 4.2) ).
05,2 "Farmakokinetické vlastnosti
Farmakokinetika sunitinibu bola hodnotená u 135 zdravých dobrovoľníkov a u 266 pacientov so solídnymi nádormi. Farmakokinetika bola podobná vo všetkých testovaných populáciách pacientov so solídnymi nádormi a u zdravých dobrovoľníkov.
V rámci dávkovacích režimov 25-100 mg sa plocha pod krivkou (AUC) a Cmax zvyšuje úmerne k dávke. Pri opakovanom dennom podávaní sa sunitinib akumuluje 3-4 krát a hlavný aktívny metabolit sa hromadí 7 až 10 krát. Rovnovážne koncentrácie sunitinibu a jeho hlavného aktívneho metabolitu sa dosahujú do 10-14 dní. Do 14. dňa sú kombinované plazmatické koncentrácie sunitinibu a jeho hlavného aktívneho metabolitu 62,9 - 101 ng / ml a predstavujú predpovedané cieľové koncentrácie na základe predklinických údajov na inhibíciu fosforylácie receptorov. in vitro a viesť k zníženiu stagnácie / rastu nádoru in vivo.
Hlavný aktívny metabolit predstavuje 23-37% celkovej expozície lieku .. Pri opakovanom podávaní jedenkrát denne alebo opakovaných liečebných cykloch v testovaných dávkovacích režimoch sa nepozorovali žiadne významné zmeny vo farmakokinetike sunitinibu alebo hlavného aktívneho metabolitu.
Absorpcia
Po perorálnom podaní sunitinibu sa maximálne koncentrácie (Cmax) spravidla pozorujú do 6-12 hodín (Tmax) od príjmu lieku.
Jedlo nemá žiadny vplyv na biologickú dostupnosť sunitinibu.
Distribúcia
Väzba sunitinibu a jeho hlavného aktívneho metabolitu na proteíny ľudskej plazmy v testoch v in vitro boli 95%, respektíve 90%, bez „zjavnej závislosti od koncentrácie.
Zdanlivý distribučný objem (Vd) sunitinibu bol veľký - 2 230 l - čo naznačuje distribúciu do tkanív.
Metabolické interakcie
Vypočítané hodnoty Ki in vitro pre všetky skúmané izoformy cytochrómu (CYP) (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 / 5 a CYP4A9 / 11) naznačili, že je nepravdepodobné, že by sunitinib a jeho hlavný aktívny metabolit v klinicky relevantnom rozsahu metabolizmus iných účinných látok, ktoré môžu byť metabolizované týmito enzýmami.
Biotransformácia
Sunitinib je metabolizovaný predovšetkým CYP3A4, izoformou cytochrómu P450, ktorá produkuje jeho hlavný aktívny metabolit, desetylsunitinib, ktorý je ďalej metabolizovaný rovnakým izoenzýmom.
Je potrebné vyhnúť sa súbežnému podávaniu sunitinibu a silných induktorov alebo inhibítorov CYP3A4, pretože plazmatické hladiny sunitinibu môžu byť zmenené (pozri časti 4.4 a 4.5).
Vylúčenie
Vylučovanie prebieha hlavne stolicou (61%) a renálna eliminácia nezmenenej účinnej látky a jej metabolitov predstavuje 16% podanej dávky.Sunitinib a jeho hlavný aktívny metabolit boli hlavnými zlúčeninami identifikovanými v plazme, moči a stolici a tvorili 91,5%, 86,4%a 73,8%v uvedenom poradí na rádioaktivite zistenej v spojených vzorkách. Drobné metabolity boli identifikované v moči a stolici, ale spravidla nie sú zistené v plazme. Celkový orálny klírens (CL / F) bol 34-62 l / h.
Po perorálnom podaní zdravým dobrovoľníkom sú polčasy eliminácie sunitinibu a jeho hlavného aktívneho metabolitu desetyl približne 40-60 hodín a 80-110 hodín.
Špeciálne populácie
Poškodenie funkcie pečeneSunitinib a jeho hlavný metabolit sú primárne metabolizované v pečeni. Systémové expozície po jednorazovej dávke sunitinibu boli podobné u subjektov s miernym alebo stredne ťažkým poškodením funkcie pečene (Childovo-Pughovo štádium A a B) v porovnaní s subjektmi s normálnou funkciou pečene. SUTENT sa neskúmal u subjektov s ťažkou poruchou funkcie pečene (Childovo-Pughovo štádium C).
Štúdie rakoviny vylúčili pacientov s ALT alebo AST> 2,5 x ULN (horná hranica normy) alebo ak boli tieto hodnoty> 5,0 x ULN v dôsledku pečeňových metastáz.
Porucha funkcie obličiek: Populačné farmakokinetické analýzy naznačili, že zdanlivý klírens sunitinibu (CL / F) nebol ovplyvnený klírensom kreatinínu v uvažovanom rozsahu (42-347 ml / min). Systémové expozície po jednej jednorazovej dávke sunitinibu boli podobné u subjektov s ťažkým poškodením funkcie obličiek ( CLcr80 ml / min). Hoci sunitinib a jeho hlavný metabolit neboli odstránené hemodialýzou u osôb s ESRD, celkové systémové expozície boli o 47% nižšie pre sunitinib a 31% pre jeho hlavný metabolit v porovnaní s osobami s normálnou funkciou obličiek.
Hmotnosť, stav výkonu: Farmakokinetické analýzy populácie naznačujú, že pre hmotnosť a výkonnostný stav skupiny Eastern Cooperative Oncology Group (ECOG) nie sú potrebné žiadne úpravy počiatočnej dávky.
Pohlavie príslušnosti: Dostupné údaje naznačujú, že ženy môžu mať o 30% nižší zdanlivý klírens sunitinibu (CL / F) ako muži; tento rozdiel však nevyžaduje úpravu počiatočnej dávky.
05.3 Predklinické údaje o bezpečnosti
Štúdie toxicity po opakovanom podávaní na potkanoch a opiciach trvajúce až 9 mesiacov ukázali, že hlavné účinky na cieľové orgány sú v gastrointestinálnom trakte (vracanie a hnačka u opíc); nadobličky (kortikálna kongescia a / alebo krvácanie u potkanov a opíc, s nekrózou, po ktorej u potkanov nasleduje fibróza); krvný a lymfatický systém (hypocelulárnosť kostnej drene a lymfoidné vyčerpanie týmusu, sleziny a lymfatických uzlín); exokrinný pankreas (degranulácia acinárnych buniek s jednobunkovou nekrózou); slinné žľazy (acinárna hypertrofia); kostné kĺby (zhrubnutie rastovej platne); maternica (atrofia) a vaječníky (zníženie vývoja folikulov). Všetky výsledky sa vyskytli pri klinicky relevantných hladinách plazmatickej expozície sunitinibu. Medzi ďalšie účinky pozorované v týchto štúdiách patrili: predĺženie intervalu QTc, zníženie ejekčnej frakcie ľavej komory (LVEF) a semenníkov tubulárna atrofia, zvýšené mezangiálne bunky v obličkách, krvácanie do gastrointestinálneho traktu a sliznice ústnej dutiny a hypertrofia predných buniek hypofýzy. endometrium) a doska na rast kostí (epifyzárne zhrubnutie alebo dysplázia chrupavky) súvisia s farmakologickým účinkom sunitinibu. Väčšina týchto udalostí bola reverzibilná po prerušení liečby na 2 až 6 týždňov.
Genotoxicita
Genotoxický potenciál sunitinibu bol hodnotený in vitro a in vivo. Sunitinib nepreukázal mutagénne účinky u baktérií využívajúcich metabolickú aktiváciu poskytovanú pečeňou potkanov. Sunitinib neindukoval štruktúrne chromozomálne aberácie v ľudských periférnych lymfocytoch in vitro. V ľudských periférnych lymfocytoch bola pozorovaná polyploidia (odchýlky v počte chromozómov) v in vitro, a to ako v prítomnosti, tak v neprítomnosti metabolickej aktivácie. Sunitinib nevykazoval žiadny klastogénny potenciál v kostnej dreni potkanov in vivo. Hlavný aktívny metabolit nebol hodnotený z hľadiska genotoxického potenciálu.
Karcinogenita
V 1-mesačnej štúdii s cieľom nájsť rozsah dávok (0, 10, 25, 75 alebo 200 mg / kg / deň), v ktorých boli transgénne myši rasH2 kŕmené orálnou sondou dávkovaním denne a kontinuálne dávkou pri podávaní v boli pozorované najvyššie dávky (200 mg / kg / deň) karcinóm a hyperplázia Brunnerových žliaz dvanástnika.
Bola vykonaná 6-mesačná štúdia na vyhodnotenie karcinogenity u transgénnych myší rasH2 kŕmených orálnou trubicou dávkovaním podanej dávky denne (0, 8, 25, 75 [znížené na 50] mg / kg / deň). Pri dávkach ≥ 25 mg / kg / deň podávaných 1 alebo 6 mesiacov (≥ 7,3 -násobok AUC pozorovaných u pacientov, ktorí dostávali odporúčanú dennú dávku [RDD]), gastroduodenálnych karcinómoch bol pozorovaný zvýšený výskyt pozadia. Hemangiosarkóm a / alebo hyperplázia žalúdočnej sliznice.
V 2-ročnej štúdii na hodnotenie karcinogenity u potkanov (0, 0,33, 1 alebo 3 mg / kg / deň), keď bol sunitinib podávaný v 28-dňových cykloch, po ktorých nasledovali 7-dňové obdobia bez podávania, sa zistil zvýšený výskyt feochromocytóm a medulárna hyperplázia nadobličiek u samcov potkanov, ktorí dostávali dávku 3 mg / kg / deň viac ako 1 rok (≥ 7,8 -násobok AUC u pacientov, ktorí dostávali RDD). Karcinóm Brunnerovej žľazy sa vyskytoval v dvanástniku v dávkach ≥ 1 mg / kg / deň u žien, pri dávkach 3 mg / kg / deň u mužov a v rovnakých dávkach bola v žalúdočných žľazách evidentná hyperplázia mukóznych buniek. deň u mužov; tieto udalosti sa vyskytli pri dávkach 0,9, 7,8 a 7,8 -násobku AUC u pacientov, ktorí dostávali RDD, v uvedenom poradí. Relevancia týchto neoplastických nálezov pozorovaných v štúdiách karcinogenity myší (rasH2 transgénnych) a potkanov pre ľudí liečených sunitinibom je neistá.
Reprodukčná a vývojová toxicita
V štúdiách reprodukčnej toxicity neboli pozorované žiadne účinky na mužskú alebo ženskú plodnosť. Štúdie toxicity po opakovanom podávaní na potkanoch a opiciach však ukázali účinky na ženskú plodnosť vo forme folikulárnej atrézie, degenerácie žltého tela, endometriálnych zmien v maternici a zníženia hmotnosti maternice a vaječníkov na klinicky významné hodnoty. systémová expozícia. Účinky na plodnosť samcov potkanov boli pozorované vo forme tubulárnej atrofie v semenníkoch, znížených spermií v nadsemenníku a koloidnej deplécie v prostate a semenných vezikulách pri plazmatických hladinách expozície 25 -násobku systémovej expozície u ľudí.
U potkanov mala embryofetálna úmrtnosť za následok významné zníženie počtu živých plodov, zvýšený počet resorpcií, zvýšené straty po implantácii a celkové straty potomstva u 8 z 28 gravidných samíc pri hladinách plazmatickej expozície 5,5-násobku systémovej expozície u ľudí. . U králikov bolo zníženie hmotnosti gravidnej maternice a počtu živých plodov spôsobené zvýšením resorpcie plodu, stratami po implantácii a celkovými stratami potomstva u 4 zo 6 gravidných samíc pri trojnásobných hladinách plazmatickej expozície. Systémová expozícia u ľudí. Liečba sunitinibom u potkanov počas organogenézy viedla k vývojovým účinkom pri dávkach ≥ 5 mg / kg / deň, ktoré pozostávali zo zvýšeného výskytu malformácií kostry plodu, charakterizovaných hlavne oneskorenou osifikáciou hrudných / bedrových stavcov a vyskytujúcich sa pri hladinách plazmatickej expozície 5,5 systémová expozícia človeka. U králikov vývojové účinky spočívali vo zvýšenom výskyte rázštepu pery pri hladinách plazmatickej expozície približne rovnakých ako hladiny pozorované na klinike a rázštepu pery a rázštepu podnebia pri hladinách plazmatickej expozície 2,7 -násobku systémovej expozície u človeka.
Účinky sunitinibu (0,3; 1,0; 3,0 mg / kg / deň) na prenatálny a postnatálny vývoj boli hodnotené v štúdii na gravidných samiciach potkanov. Prírastok telesnej hmotnosti matky počas gravidity a laktácie sa znížil pri dávkach> 1 mg / kg / deň, ale až do dávok 3 mg / kg / deň (odhadovaná expozícia> 2, 3 -násobok AUC pozorovaná u pacientok sa nepozorovala žiadna reprodukčná toxicita pre matku). príjem RDD) Znížené telesné hmotnosti potomstva boli pozorované pri dávkach 3 mg / kg / deň v období pred a po odstavení.Pri dávkach 1 mg / kg / deň (expozícia približne ≥0,9 -násobok AUC pozorovaná u pacientov, ktorí dostávali RDD) nebola pozorovaná žiadna vývojová toxicita.
06.0 FARMACEUTICKÉ INFORMÁCIE
06.1 Pomocné látky
Obsah kapsuly
Manitol (E421)
Sodná soľ kroskarmelózy
Povidón (K-25)
Stearan horečnatý
Obal kapsuly
Želé
Červený oxid železitý (E172)
Oxid titaničitý (E171)
Atrament
Šelak
Propylénglykol
Hydroxid sodný
Povidón
Oxid titaničitý (E171)
06.2 Nekompatibilita
Nie je to relevantné.
06.3 Obdobie platnosti
3 roky.
06.4 Špeciálne opatrenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
06.5 Charakter vnútorného obalu a obsahu balenia
Fľaše z polyetylénu s vysokou hustotou (HDPE), s polypropylénovým uzáverom, obsahujúce 30 tvrdých kapsúl.
Priehľadné poly (chlórtrifluóretylénové) / PVC blistre s perforovanými jednotlivými dávkami a s hliníkovou fóliou s tepelne zatavovacím lakom obsahujúcim 28 x 1 tvrdých kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
06.6 Návod na použitie a zaobchádzanie
Žiadne špeciálne pokyny.
07.0 DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Ltd.
Ramsgate Road
Sendvič, Kent CT13 9NJ
UK
08.0 REGISTRAČNÉ ČÍSLO
EU/1/06/347/001
037192022
EU/1/06/347/004
09.0 DÁTUM PRVEJ REGISTRÁCIE ALEBO OBNOVENIA REGISTRÁCIE
Dátum prvej registrácie: 19. júla 2006
Dátum posledného obnovenia: 9. januára 2012
10.0 DÁTUM REVÍZIE TEXTU
11.0 PRE RÁDIOVÉ DROGY, ÚPLNÉ ÚDAJE O VNÚTORNEJ DOSIMETRII ŽIARENIA
12.0 PRE RÁDIOVÉ DROGY, DODATOČNÉ PODROBNÉ NÁVODY NA PRÍPADNÚ PRÍPRAVU A KONTROLU KVALITY